



¿ Introducción
La cistinosis es un trastorno metabólico raro con herencia autosómica recesiva, que se caracteriza por la acumulación del aminoácido cistina dentro de los lisosomas,1 con un depósito tisular generalizado de cristales de cistina no proteica, como resultado de un defecto en el transporte lisosomal en células de hígado, bazo, ganglios linfáticos y médula ósea, pudiéndose también depositar en el riñón y el ojo.2
Entre los sinónimos con los cuales se conoce a esta enfermedad en la forma infantil están cistinosis nefropática infantil, enfermedad de Fanconi, enfermedad de Toni-Fanconi Lignac, forma adolescente y forma adulta.
La forma infantil se presenta en uno de cada 200 000 individuos y suele detectarse entre los cuatro y 12 meses de edad, es más común entre los francocanadienses; sin embargo, no se ha descrito ninguna predisposición por sexos, la forma adolescente, en la segunda década y la forma adulta, desde los 15 hasta los 50 años.3
Clasificación:3 Forma infantil o nefropática: se caracteriza por enanismo y disfunción renal progresiva, con depósito de cristales policromáticos finos de cistina en conjuntiva, estroma corneal y otras partes del ojo; siempre es mortal en la primera década de la vida, si no se realiza trasplante renal.
1. Forma intermedia o del adolescente: tiene una afectación renal menos grave, produciéndose la muerte en la segunda o tercera décadas de la vida.
2. Forma adulta: la esperanza de vida es normal.1
Cuadro clínico: Las manifestaciones sistémicas son características, consistiendo en: presencia de poliuria, dificultad para ganar peso, retraso del crecimiento, raquitismo, fiebre inexplicable, insuficiencia renal y fotofobia, siendo más grave y de aparición más precoz en la forma infantil y no se asocia a retraso mental. En las formas juveniles y adultas el comienzo es más tardío y la evolución más leve.
Manifestaciones oculares: En todas las formas pueden aparecer depósitos cristalinos en forma de aguja en la córnea, la conjuntiva y el iris. En la córnea central los cristales se localizan a nivel del estroma superficial. En la periferia son más densos y están dispersos por todo el espesor estromal.
Las anomalías pigmentarias retinianas son exclusivas de la variante infantil de cistinosis; empiezan en la periferia, aunque pueden terminar por afectarle completamente.
Diagnóstico: Éste se realiza primordialmente de forma clínica por hallazgos corneales y conjuntivales. Pueden determinarse las concentraciones de cistina en leucocitos o fibroblastos cultivados. La angiografía con fluoresceína puede descubrir defectos en ventana en el epitelio pigmentario de la retina.
Se puede realizar biopsia conjuntival la cual mostraría los típicos depósitos cristalinos.
Es posible detectar la forma infantil en diagnóstico prenatal si se demuestra un contenido elevado de cistina en células de líquido amniótico.
Tratamiento: Médico: el tratamiento sistémico con cisteamina (mercaptamina) ayuda a reducir el exceso de cistina, prolongando la vida y retrasando la aparición de la insuficiencia renal. Puede ser necesario un tratamiento con cisteamina tópica para aliviar los síntomas oculares como la fotofobia.3 La cisteamina tópica reacciona con la cistina intracelular, formando un disulfuro de cisteinacisteamina que se parece a la lisina y se transporta a través del lisosoma utilizando el sistema de transporte normal de la lisina. Se ha observado la recurrencia de los cristales en pacientes sometidos a una queratoplastía penetrante por la turbidez asociada a los depósitos corneales. Estos cristales policromáticos refractivos son más densos en la zona periférica de la córnea pero se ven en todo el estroma anterior, incluso dentro de la zona central de la córnea. La forma adulta no suele requerir tratamiento.1
1. Quirúrgico: el trasplante renal suele prolongar la supervivencia.3
¿ Presentación del caso
Hombre de 14 años de edad, asintomático, sin antecedentes heredofamiliares o personales patológicos, quien fue internado en un hospital del sector salud por haberse detectado albuminuria en examen general de orina rutinario, de seis meses de evolución, siendo evidente albuminuria, glucosuria y proteínas en orina de 24 horas de 10.3 g. Otros paraclínicos demostraron, creatinina sérica de 2.5 mg/100 mL, nitrógeno ureico 37 mg/100 mL, colesterol sérico total 277 mg/100 ml, hematocrito de 48, velocidad de sedimentación globular de 33 y bajos niveles de albúmina sérica. También se determinaron anticuerpos anti desoxiribonucleoproteínas y la fracción B1-C de complemento determinado por inmunodifusión dentro de límites normales. La pielografía excretora fue de características normales.
Se llevó a cabo biopsia renal en la que se informó glomerulonefritis proliferativa con datos de hialinización y nefritis por complejos inmunes en la membrana basal glomerular determinados por inmunofluorescencia. Un año después de su primer ingreso fue admitido en el servicio de pediatría del Hospital General de México por detectarse disminución en la depuración de creatinina, razón por la que se inició hemodiálisis.
En la esfera ocular, el paciente manifestó la presencia de "mancha" en el campo visual del ojo derecho razón por la que se solicitó valoración por el servicio de oftalmología.
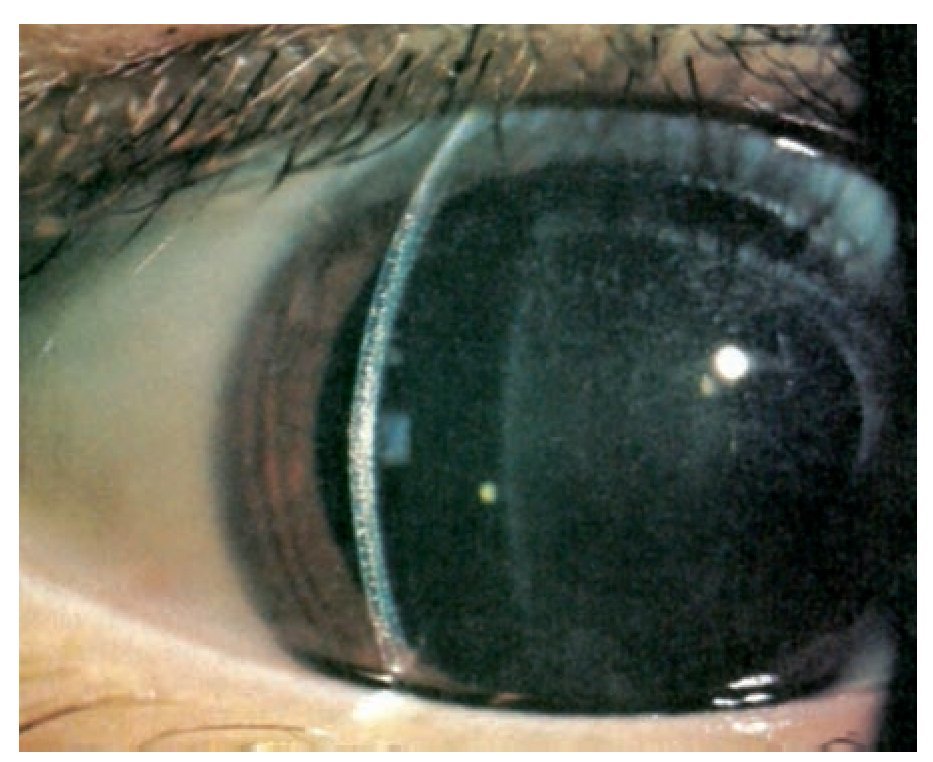
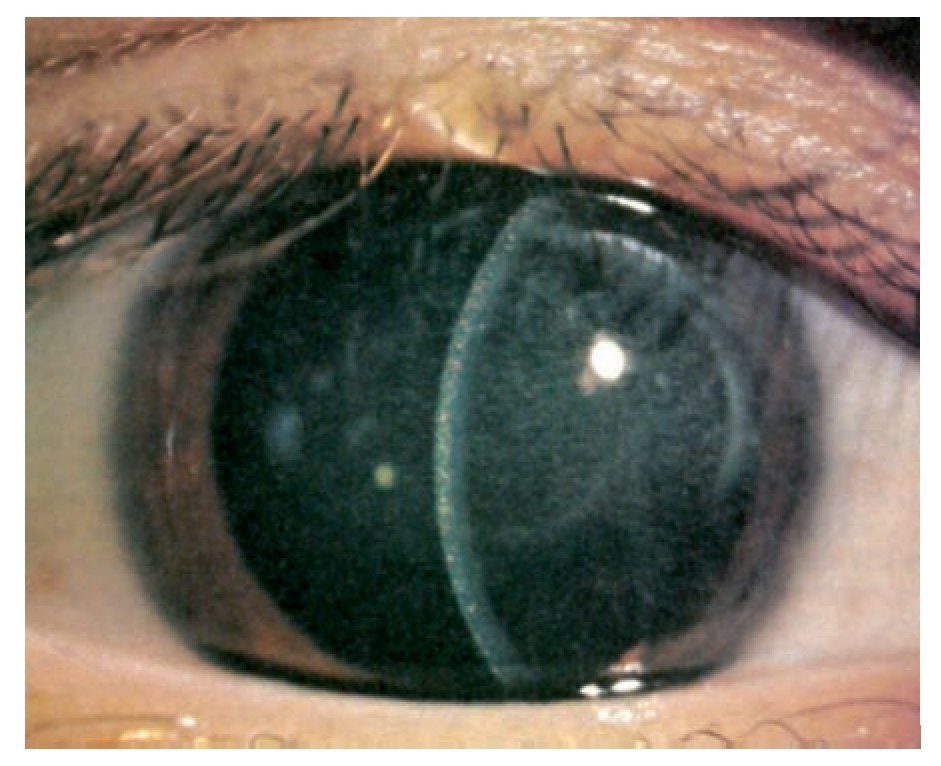
En la exploración ocular se determinó una AV OD 20/400 y OI 20/25 con PIO de 13 mmHg en ambos ojos. Bio-microscópicamente en el ojo derecho se encontró, en la córnea y conjuntiva la presencia de múltiples cristales puntiformes localizados en las 2/3 partes del estroma anterior (Figuras 1 y 2) y con la fundoscopia se observó una hemorragia prerretiniana en el área macular del ojo derecho. El ojo izquierdo fue completamente normal.
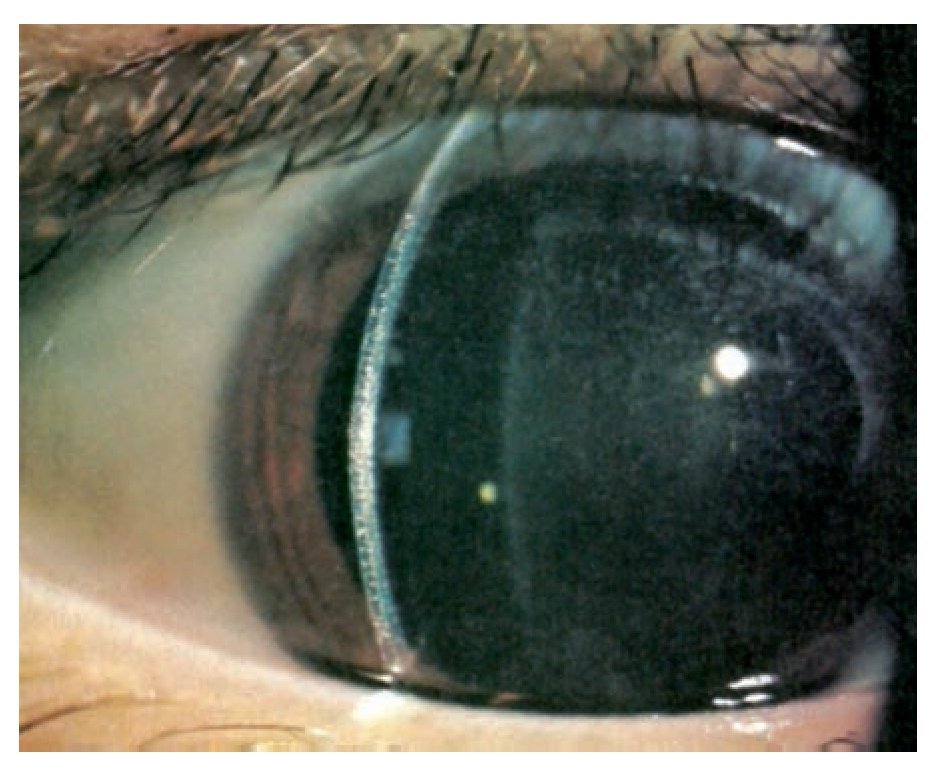
¿ Figura 1. Biomicroscopía de globo ocular derecho, córnea periférica, en que se observan múltiples depósitos localizados en el espesor total del estroma corneal.
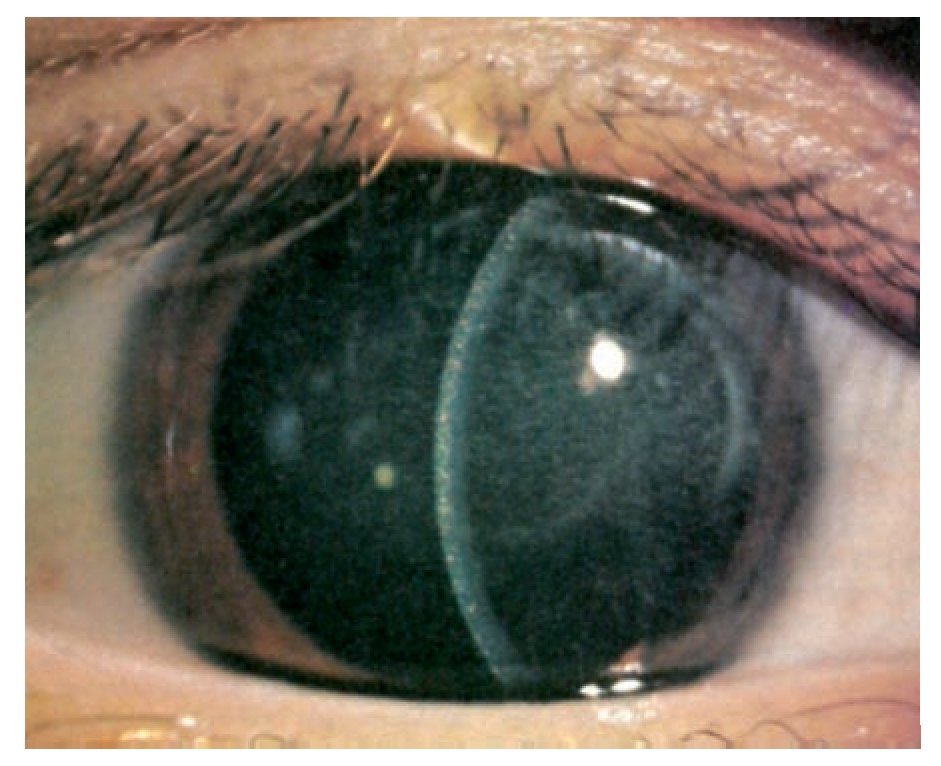
¿ Figura 2. Biomicroscopía de ojo derecho de córnea central en que es evidente la presencia de múltiples depósitos en los 2/3 anteriores del estroma corneal.
Se planteó la posibilidad de trasplante renal mismo que no se llevó a cabo por abandono de tratamiento desconociéndose el estado actual del paciente.
¿ Discusión
La cistinosis nefropática infantil se caracteriza por el depósito exageradamente elevado de cistina libre en diferentes órganos y tejidos, debido a un defecto en el transporte de cistina a través de la membrana lisosomal.1-4 Es una enfermedad autosómica recesiva, de rara ocurrencia, que afecta de 1/100 000 a 1/200 000 niños.5 Ambos padres suelen ser heterocigotos y contienen aproximadamente cinco veces la cantidad normal de cistina libre intracelular, si bien ellos son clínicamente sanos con riesgo de recurrencia en los hermanos de un sujeto afectado de 25% en la literatura revisada,6 no habiéndose encontrado información sobre la incidencia de este padecimiento en la población latina o mexicana. En el caso presentado, no fue posible determinar el tipo de herencia debido a la inasistencia para la realización de diferentes estudios, incluyendo el genético, aunque se infiere que tuvo una variedad intermedia por la edad de presentación, a diferencia del reporte de caso donde Gahl y colaboradores encontraron un caso con la forma adulta.6
En 1967 se demostró con microscopía electrónica la alteración lisosomal en esta patología y en 1980 con radioisótopos, la de su transportador,7 y cabe mencionar que el grupo colaborativo de Investigación en cistinosis mapeó el gen específico en el brazo corto del cromosoma 142 corroborado por estudios de ligamiento.8-9
La cistina es un derivado disulfurado formado por la oxidación de dos moléculas de cisteína, que en la cistinosis se depositan en el interior de los lisosomas en forma de cristales10 siendo, por lo tanto, un fenómeno intracelular con niveles plasmáticos normales de cistina.11 Se han identificado cristales de cistina en diversos tejidos como riñón, córnea, páncreas, hígado, pulmón, intestino, bazo, médula ósea, músculos, sistema nervioso central, tiroides.12-13 Esta patología progresiva conduce a fallas de estos parénquimas, determinando tubulopatía14 e insuficiencia renal, y la alteración ocular reportada en el presente caso sin la coexistencia con hipotiroidismo, diabetes, hipertensión portal, hiperesplenismo, miopatía, deterioro intelectual y otros previamente reportados.15-17
En relación al diagnóstico diferencial, importa hacer notar que la causa más frecuente de síndrome de Fanconi en el niño es la cistinosis.15 Deben destacarse otras etiologías en presencia de este síndrome, como la tirosinemia, galactosemia, intolerancia a la fructosa, enfermedad de Wilson, síndrome de Lowe, glucogenosis (Tabla 1) y la intoxicación por metales pesados y glucoglicinuria, mismas que no se diagnosticaron en este caso de cistinosis en adolescente.

Los cristales corneales se presentan de múltiples formas de aguja, opacidades altamente reflectivas valorables fácilmente por biomicroscopía con lámpara de hendidura, presentes en el epitelio corneal, estroma y endotelio.17 Debido a su aspecto característico y distribución son fácilmente reconocibles, pudiendo diferenciarse de otras queratopatías cristalinas. La acumulación de cristales en la córnea se inicia en la infancia y es, sin duda, evidente desde los 16 meses de edad;4 ésta se inicia en la periferia anterior y avanza posteriormente de forma centrípeta. Desde aproximadamente los siete años de edad, toda la periferia del estroma y el endotelio acumulan cristales hasta que a los 20 años dichos cristales se pueden ver en todo el estroma corneal.17,18 En este caso, a pesar de ser aún menor de edad, se observó por bio-microscopía como múltiples cristales puntiformes localizados en las dos terceras partes del estroma anterior corneal periférico. El aumento de la densidad de los cristales de cistina en la córnea resulta en calina corneal que es fácilmente reconocible a simple vista, fenómeno no observado en este caso.
Los cristales corneales son inicialmente asintomáticos, pero la fotofobia puede desarrollarse en los primeros años de vida6 y la gravedad de ésta varía, por lo que muchos pacientes requieren anteojos oscuros y otros poseen un blefaroespasmo significativo.4,6 Se observa ocasionalmente queratopatía punteada superficial asociada con sensación de cuerpo extraño y dolor sobre todo en pacientes mayores de 10 años de edad.18
Kaiser Fujikawa y colaboradores17 han documentado pérdida de sensibilidad al contraste, aumento del glare, disminución de la sensibilidad corneal y aumento del grosor corneal en pacientes con cistinosis nefropática, especulándose que estos cambios son el resultado de los depósitos de cristales en la córnea; sin embargo, se ha comprobado que los cristales no afectan la agudeza visual, por lo que si ésta disminuye debe conducir a la investigación de otra etiología subyacente. En este caso, se encontró que la causa de la disminución de la agudeza visual fue por la hemorragia prerretiniana en el área macular, cuya etiología puede haber resultado de un aumento agudo de las cifras de tensión arterial sistémica por la nefropatía o al depósito de cristales en la microvasculatura, lo que determinó un fenómeno oclusivo, a diferencia de un caso reportado previamente en que la baja de visión fue secundaria a una membrana neovascular macular.18
Los cristales también se han encontrado en la cámara anterior, iris y cuerpo ciliar,4 coroides, y el nervio óptico4,6,17 pero no se observaron en el caso reportado. En algunos pacientes se ha determinado una retinopatía pigmentaria. Este hallazgo precede a la aparición de los cristales de la córnea y se ha visto tan temprano como a la edad de cinco semanas; los cambios pueden estar presentes desde la vida fetal. La retinopatía pigmentaria; sin embargo, no parece ser un hallazgo constante siendo más comúnmente descritas las zonas de despigmentación con zonas moteadas de pigmentación.6 La alteración pigmentaria se limita a la periferia en las primeras etapas, más en el sector temporal que en los segmentos nasales y aparecen bilaterales y simétricos, mas este paciente tampoco los presentó. Se han descrito anomalías maculares desde los seis años de edad.18
Desde el inicio del trasplante renal y con el éxito de la terapia los pacientes con cistinosis viven más tiempo, lo que manifiesta las complicaciones a largo plazo de la enfermedad en múltiples órganos y sistemas. La experiencia inicial con los pacientes operados de trasplante corneal no reveló problemas oculares importantes.4 Sin embargo, con un seguimiento más largo, se ha informado de mayores complicaciones del segmento anterior, queratopatia punteada superficial y queratopatia filamentosa, sinequias del iris así como anomalías en el ángulo.18
Los primeros casos de cistinosis se diagnosticaron por la presencia de cristales de cistina en la córnea17 y médula ósea. Actualmente es posible medir el contenido de cistina en el interior de los leucocitos, lo que requiere una preparación especial de la muestra sanguínea, para separar los glóbulos rojos. Los leucocitos son lisados y precipitados en medio ácido, determinándose el contenido de cistina por una técnica de dilución isotópica, utilizando una proteína especifica que liga la cistina.19
Uno de los mayores avances lo constituye el tratamiento con cisteamina, medicamento que ha sido recientemente aprobado para su uso por la FDA. Es una thiolamina utilizada oralmente como reductor de la cistina en la cistinosis que pasa a través de las membranas de los lisosomas interactuando con la cistina para formar cisteína y una mezcla de cisteaminacisteína disulfido. La cisteína atraviesa las membranas lisosómicas, utilizando el sistema transportador para lisina. Luego del tratamiento con cisteamina se ha reportado depleción significativa de cistina en los leucocitos polimorfonucleares; también en el músculo esquelético, hígado, riñón, páncreas, pulmón y bazo.19
La administración sistemática de cisteamina no altera el depósito de cristales de cistina en la córnea.19,20
La iniciación precoz del tratamiento con cisteamina puede prevenir el deterioro renal y evitar el trasplante, cambiando radicalmente el pronóstico de la enfermedad y mejorando el crecimiento pondoestatural durante la infancia; la terapia permanente también puede prevenir las complicaciones no renales de la cistinosis. Cuando se ha hecho el diagnóstico del síndrome de Fanconi, la administración de cisteamina no mejora el daño renal ya instalado ni la tubulopatía. Esta última podría ser prevenible si se iniciara el tratamiento en las primeras semanas de la vida.21
Uno de los enigmas aún no resueltos en la cistinosis es el completo conocimiento de los mecanismos de daño celular producidos por la acumulación de cistina. La cistina bien aislada en el interior de los lisosomas no debería ser tóxica, así como el disulfuro de cisteamina y cisteína, una vez en el citoplasma donde el estado de óxido-reducción está controlado por el ciclo del glutatión. Algunos datos recientes sugieren una alteración de los mecanismos de apoptosis en las células cistinóticas. Queda pendiente determinar si los diversos grados de apoptosis pueden estar en relación con las diversas expresiones fenotípicas de la enfermedad en población latina así como en la repercusión ocular de este padecimiento, motivo sin duda de futuras investigaciones en el área de la bioquímica molecular.
Correspondencia: Dr. Ariel Prado Serrano.
Dr. Balmis N° 144 Col. Doctores. Delegación Cuauhtémoc, México D. F.
Teléfono: 5004 3801.
Correo electrónico: ariprase@hotmail.com, Ariuiamx@yahoo.com
