


El meduloepitelioma es un tumor muy poco frecuente en adultos, encontrándose descritos en la literatura solo una decena de casos. Se presenta la historia de un paciente sometido a enucleación de su ojo derecho por sospecha de melanoma coroideo. La biopsia informó neoplasia de estirpe neural de comportamiento biológico incierto, y el estudio inmunohistoquímico informó neoplasia neuroendocrina de bajo grado. Se realizó estudio de extensión sistémica que resultó negativo. El paciente no se somete a controles durante 6 años, volviendo a consultar por una gran ocupación orbitaria derecha, manejada mediante exenteración orbitaria. La biopsia informó neoplasia maligna de estirpe neural, y la histoquímica concluyó meduloepitelioma pigmentado maligno ocular con extensión a la órbita. El paciente evolucionó con signos de remisión completa de la lesión, sin embargo un año y medio después comenzó con un aumento de volumen rápidamente progresivo, que compromete toda la región orbitaria derecha. Se presenta el caso a comité oncológico, indicando estudio de extensión sistémica mediante citología de LCR, TAC de tórax, abdomen y pelvis que resultaron normales; el cintigrama óseo evidenció actividad osteoblástica patológica en macizo facial, y la RMN de cerebro evidenció invasión intra y extracraneal, con ocupación de los senos paranasales. Con estos antecedentes se le ingresó en el programa de cuidados paliativos y alivio del dolor, realizándose radioterapia paliativa, con remisión parcial de tamaño tumoral y sintomatología.
The medulloepithelioma is a tumor too little frequent in adults, meeting described in literature only a group of ten of cases. We presented the case of patient submitted to enucleation of its straight eye by suspicion of melanoma uveal. The biopsy informed neoplasia of neural lineage of biological uncertain behavior, and the study immunohistochemistry informed neoplasia neuroendocrina of low grade. Study of systemic extension proved to be negative. The patient loses controls for 6 years, returning to consult for a great orbital right occupation, treated with orbital exenteration. The biopsy informed neoplasia malignant of neural lineage, and the histochemistry informed the pigmented malignant ocular medulloepithelioma with extension to the orbit. The patient evolved with the lesion's signs of complete remission, however a year and six months after begin with an increase of rapidly progressive volume, occupying all of the orbital region. Oncologic committee indicate study of systemic extension with LCR‘s cytology, TAC of thorax, abdomen and pelvis that proved to be normal; the osseous cintigrama evidenced activity osteoblastic pathological in facial region, and MRI of brain evidenced invasion intra and extra-cranial, with occupation of the paranasal sinus. The patient enters to the program of palliative cares and relief of the pain, receiving palliative radiation therapy, with partial remission of size tumoral and symptomatology.
El meduloepitelioma es un tumor congénito poco frecuente, originado en el neuroepitelio embrionario antes de su diferenciación hacia epitelio no pigmentado del cuerpo ciliar1–10. Este tejido embrionario reviste la cavidad del tubo neural primitivo y la vesícula óptica, por lo que el tumor puede localizarse además de en el cuerpo ciliar (lugar de mayor frecuencia), en otros órganos derivados del neuroepitelio embrionario como la retina, papila, nervio óptico e iris1–3,11,12. Puede localizarse también en el sistema nervioso central, donde es generalmente periventricular con afectación de los lóbulos temporal, parietal, occipital y frontal, por orden de frecuencia13.
Se presenta generalmente en la infancia, con una edad media a los 5 años, siendo en extremo raros los casos en adultos1,4,5,8,14, con una edad media al diagnóstico de 45 años1. Una revisión de casos efectuada el 2011 daba cuenta solo de 12 casos publicados hasta esa fecha1. Habitualmente es unilateral y focal, aunque hay descritos casos bilaterales4,6,10, y se presenta con disminución de la agudeza visual, epífora, dolor, rubeosis iridiana, leucocoria, exoftalmos, estrabismo, subluxación del cristalino, catarata y masa en el iris, cuerpo ciliar o cámara anterior1,3–5,8,9,14. Algunos autores consideran que la presencia de coloboma zónulo cristaliniano congénito, membrana inflamatoria pre o retrolental («seudomembrana ciclítica») y el glaucoma neovascular con ausencia de alteraciones retinianas es patognomónica de meduloepitelioma4,7–9. El tumor produciría una lesión isquémica por acción mecánica, con neovascularización subsecuente y oclusión del ángulo anterior, produciendo de esta forma la hipertensión ocular y glaucoma.
Histológicamente se clasifican en teratoides y no teratoides (considerando la presencia o no de un componente heterotópico como cartílago, músculo estriado o tejido cerebral), y a su vez en malignos y benignos. La inmunohistoquímica muestra la diferenciación existente hacia distintas líneas celulares1–4,8,9,15.
Caso clínicoPaciente masculino, de 57 años, de nacionalidad boliviana, sin antecedentes mórbidos, que el año 2005 consulta en policlínico por un ojo ciego derecho, con gran tumoración grisácea, con rotura escleral espontánea por donde protruye contenido uveal, sin permitir distinguir estructuras internas oculares, de 3 años de evolución. Se lleva a cabo una ecografía ocular que informa «un globo ocular ocupado casi en su totalidad por una masa tumoral abollonada e irregular, que hace cuerpo con la pared posterior, con estructura acústica heterogénea y reflectividad interna intermedia a alta, con compromiso escleral posterior y superior, siendo estos hallazgos compatibles con un extenso melanoma necrótico, con protrusión extraocular». Con estos antecedentes el paciente fue sometido a una enucleación de su ojo derecho, describiendo en el protocolo operatorio «un gran tumor que compromete el tercio superior de la esclera, la que prácticamente ha desaparecido, con rotura escleral superior». La biopsia informó «fragmentos de globo ocular con infiltración por neoplasia de disposición trabecular, seudoglandular, en partes organoides, principalmente de estructuras anteriores: plexos coroideos, cuerpo ciliar, iris y córnea. Con mínima tasa mitótica, sin áreas de necrosis», concluyendo una «neoplasia de estirpe neural de comportamiento biológico incierto». El patólogo refiere que se podría tratar de un meduloepitelioma coroideo del adulto. Se realizó estudio inmunohistoquímico cuyos resultados fueron «cromogranina A negativa en células neoplásicas, pancitoqueratina positiva en aproximadamente el 20% de las células neoplásicas, S-100 positiva en aproximadamente el 40% de las células neoplásicas», apoyando la presencia de una neoplasia neuroendocrina de bajo grado. Con estos antecedentes se presentó el caso al comité oncológico, realizándose estudio con TAC de cuello y tórax, y ecografía abdominal, encontrando solo imagen sugerente de adenopatía axilar izquierda, que fue biopsiada sin reconocer tejido maligno.
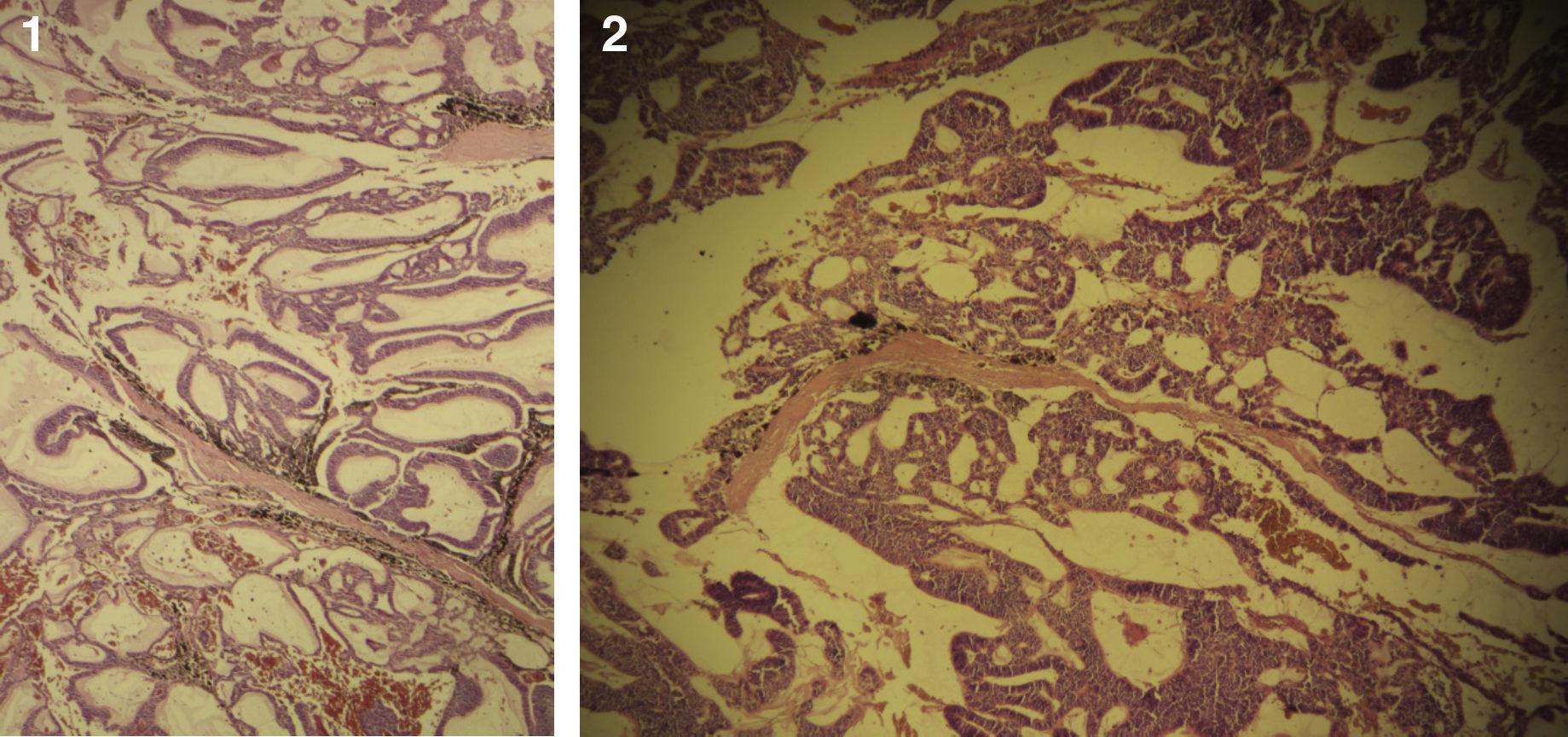
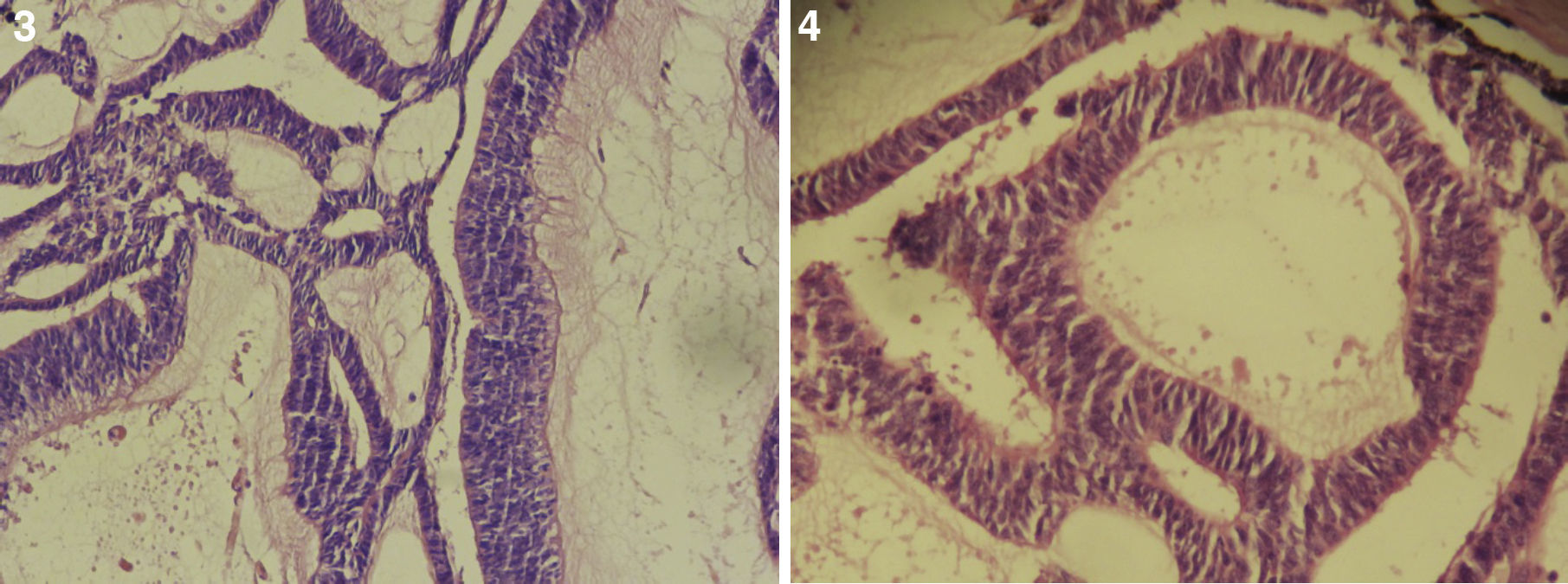
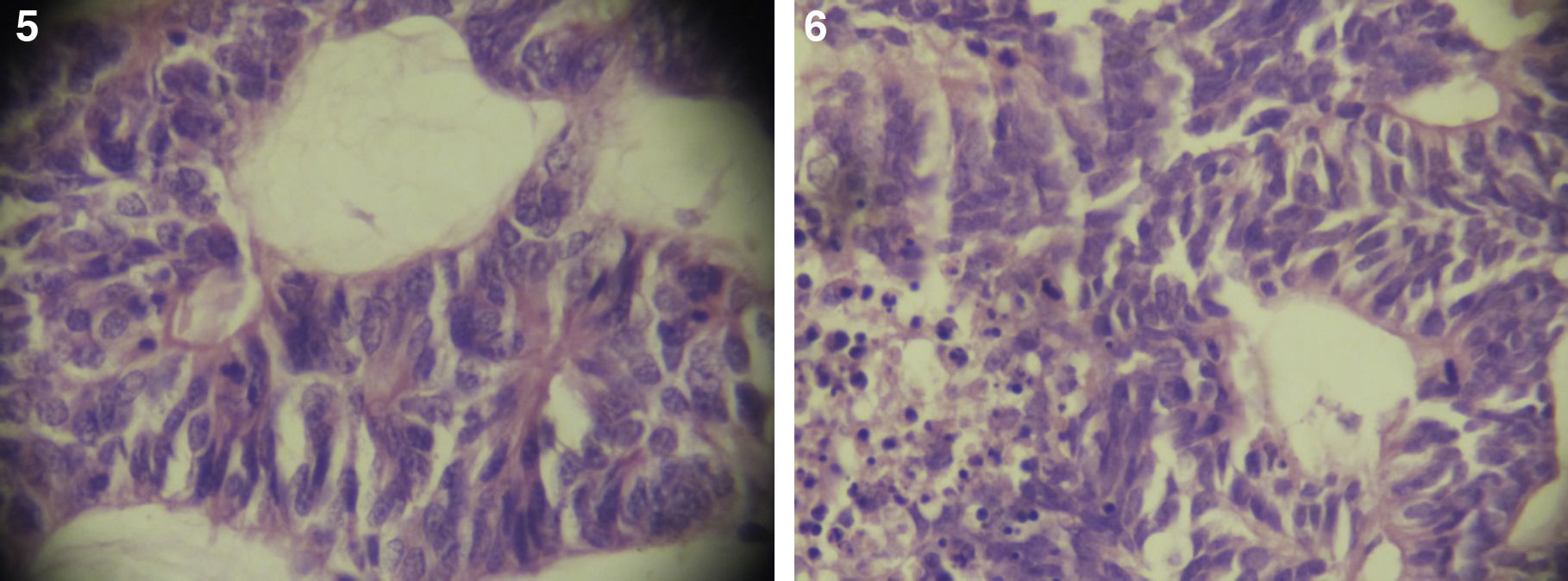
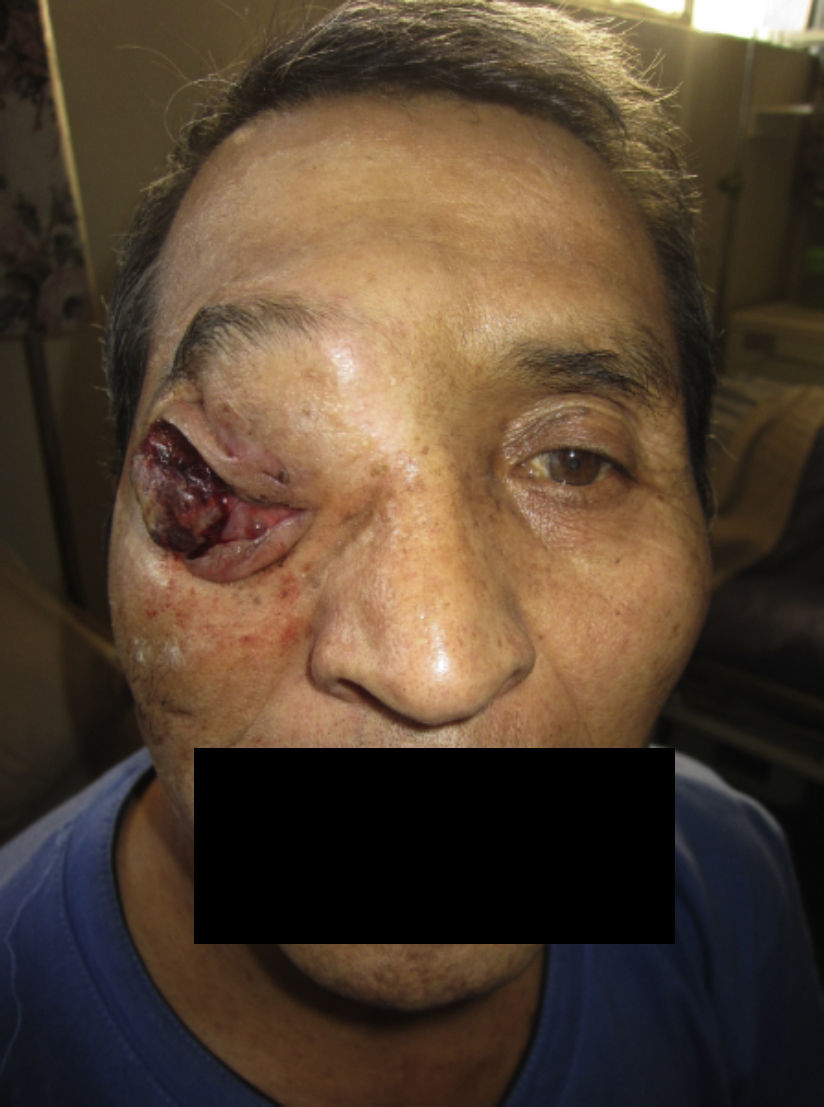
El paciente no se somete a controles durante 6 años, volviendo a consultar por gran ocupación de toda la órbita derecha hasta su reborde, sin compromiso de párpados, blanda, de consistencia quística, indolora, de 2 meses de evolución. Se hace TAC de órbitas que evidencia una lesión tumoral heterogénea, lobulada, que llena la órbita derecha, sin comprometer sus paredes. Se realizó una exenteración orbitaria, respetando los párpados como manejo. La biopsia del tejido removido informó «formación aproximadamente ovoidea, ocupada completamente por tumor redondeado, grisáceo, con estrías negruzcas, con lesión satélite hacia uno de los extremos; hallazgos histológicos compatibles con neoplasia maligna de estirpe neural con compromiso del margen quirúrgico». La histoquímica informó «proliferación neoplásica de hábito epitelial, dispuesta en trabéculas, dejando cavidades irregulares; el epitelio cilíndrico es seudoestratificado, sin evidencia de actividad secretora, con desarrollo de numerosas rosetas de tipo neuroepitelial; una proporción de las células presentan pigmentación; hay discreta actividad mitótica» (figs. 1–6). «Vimentina positivo intenso en células neoplásicas, patrón citoplasmático. Proteína S-100 positivo intenso en células neoplásicas, patrón nuclear y citoplasmático. Cromogranina positivo discreto en células neoplásicas, patrón citoplasmático. Sinaptofisina, TTF1, melan A, proteína acídica fibrilar glial y citoqueratina de alto y bajo peso molecular (AE1- AE3) negativos en células neoplásicas. Hallazgos concordantes con un meduloepitelioma pigmentado maligno ocular con extensión a la órbita».



Cuatro meses después se le realizó una limpieza quirúrgica por una gasa retenida en la cavidad ocular, que no se había retirado al no asistir el paciente a los controles postoperatorios, enviando a estudio muestras de tejido de mucosa etmoidal y de granulación, sin reconocer indicios de tumor maligno.
El paciente evolucionó de manera favorable, por lo que un año más tarde se realizó un injerto dermograso tomado de fosa ilíaca derecha, con el fin de reformar sus fondos de sacos y permitir el implante de una prótesis ocular.
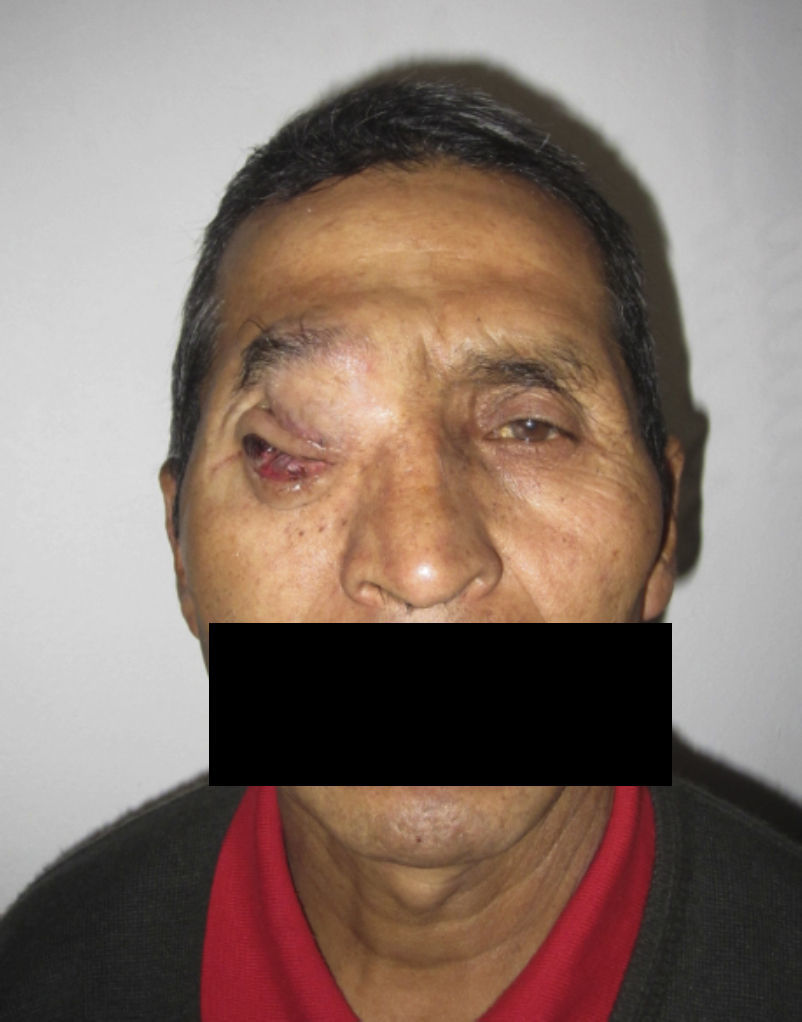
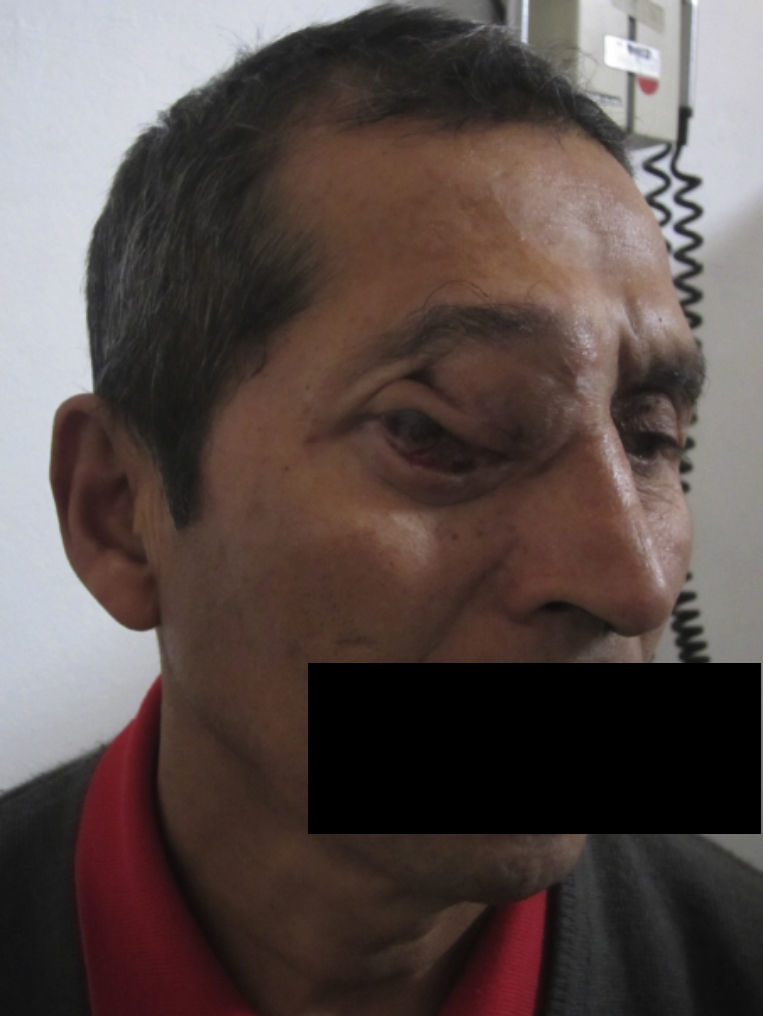
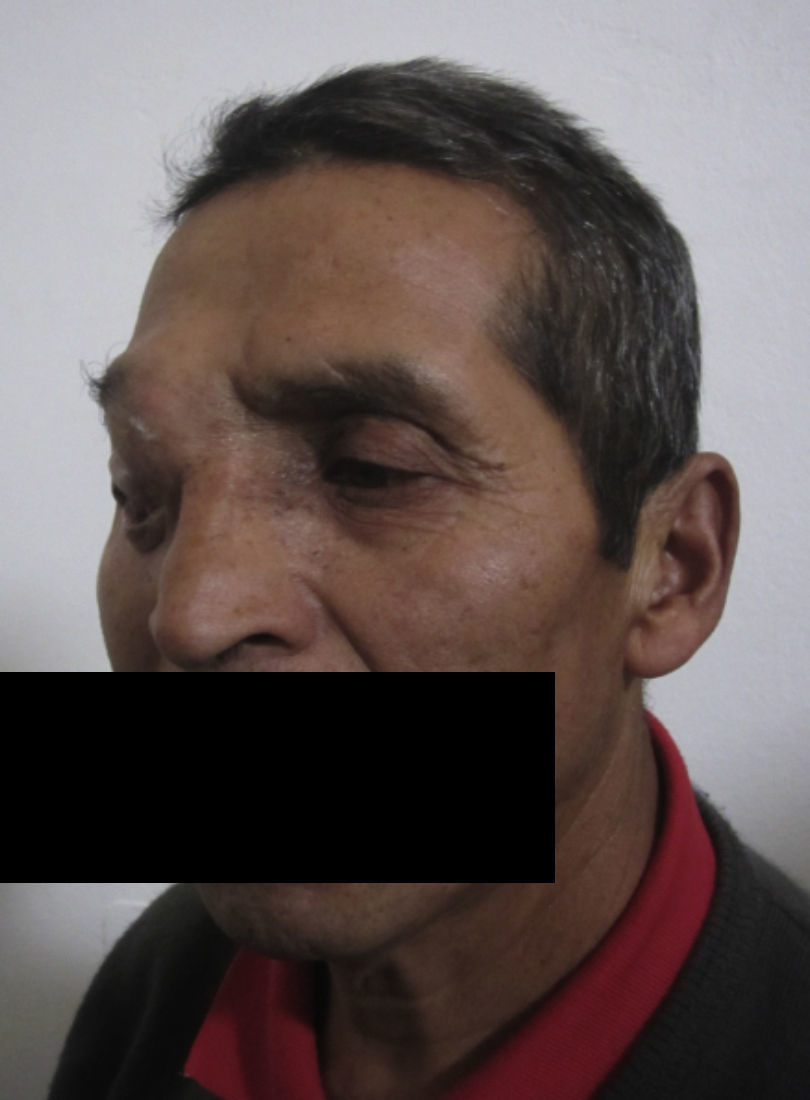
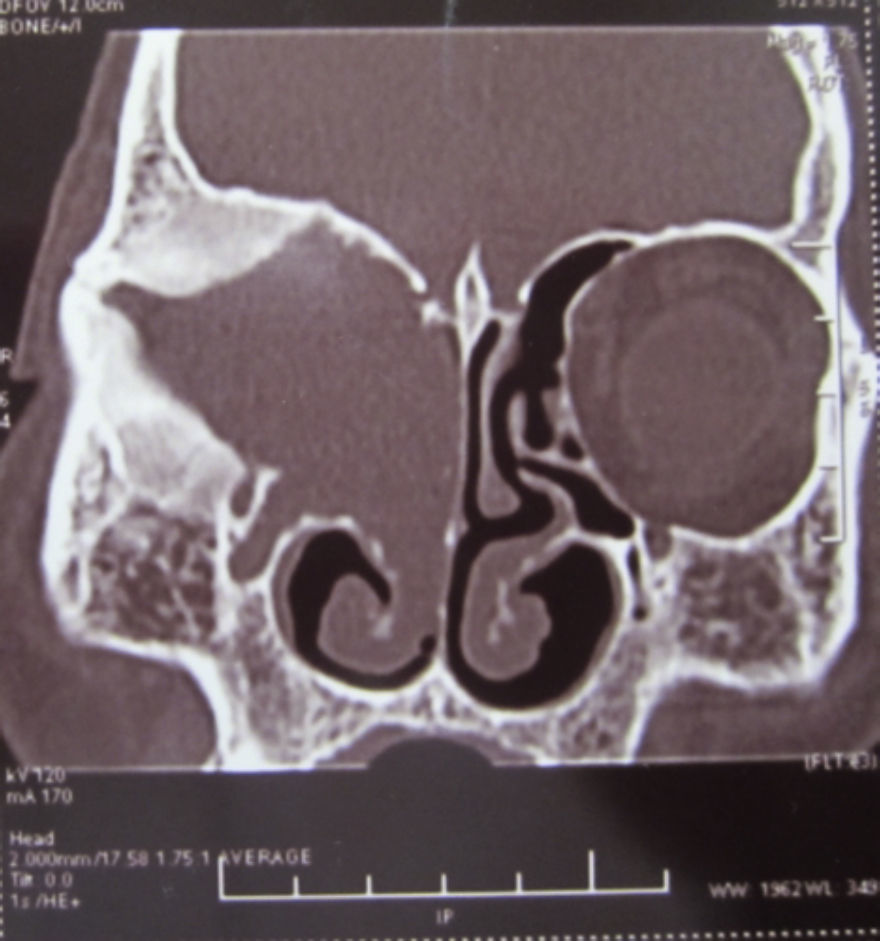
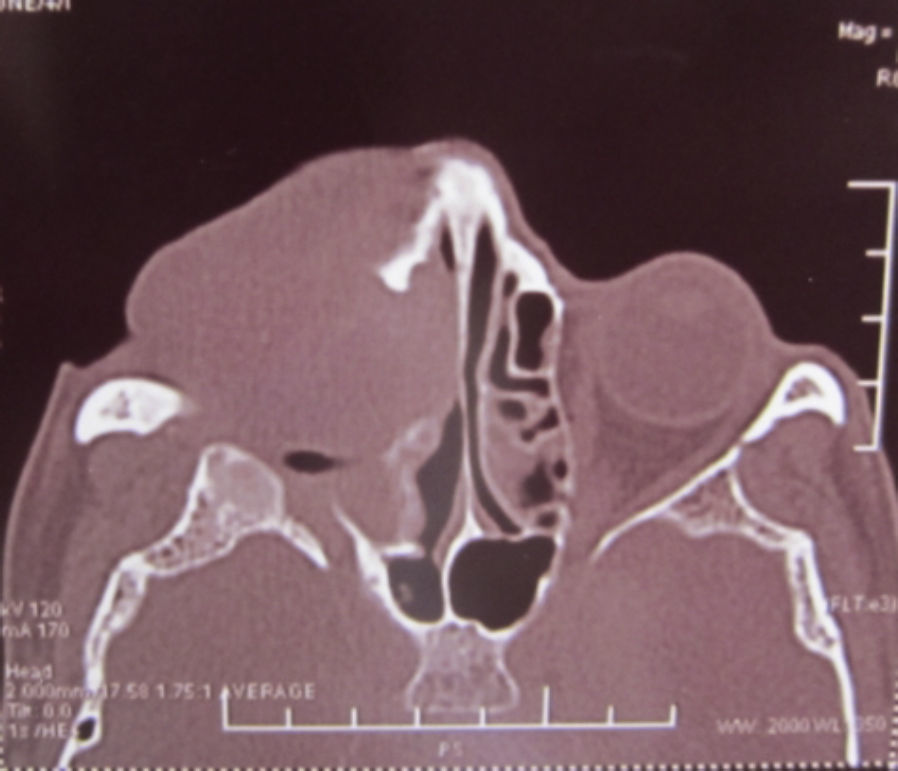
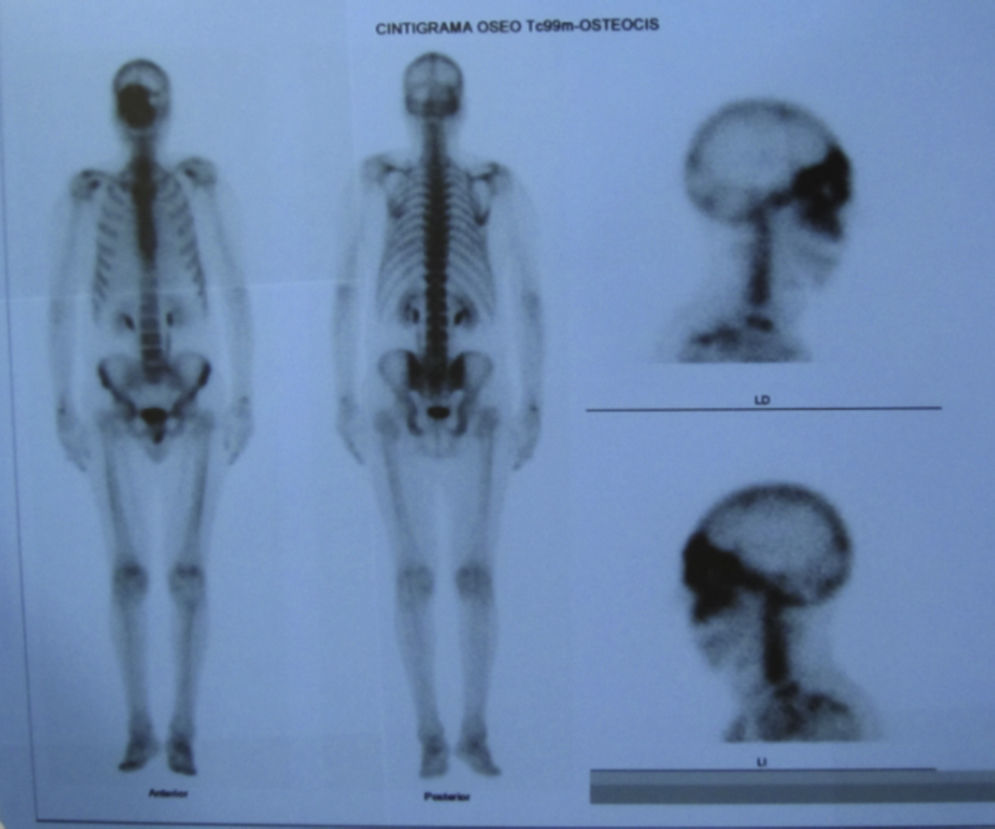
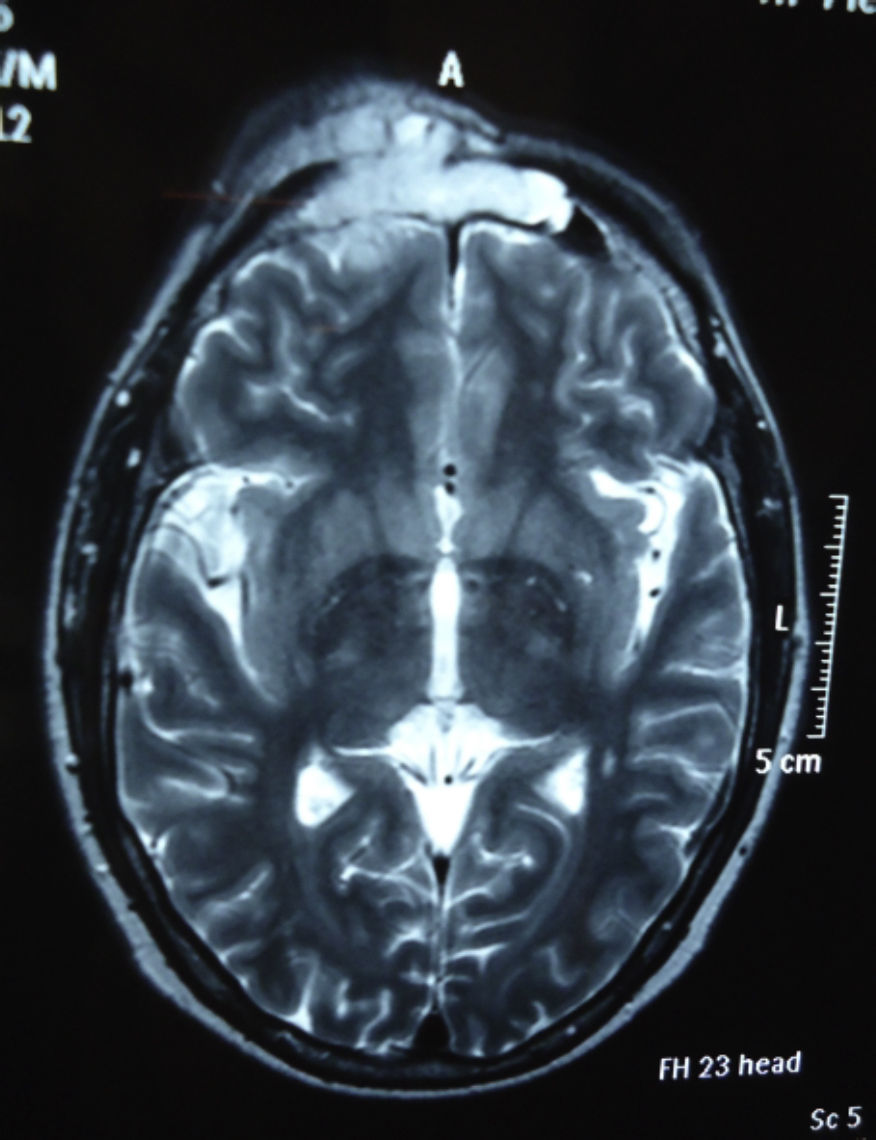
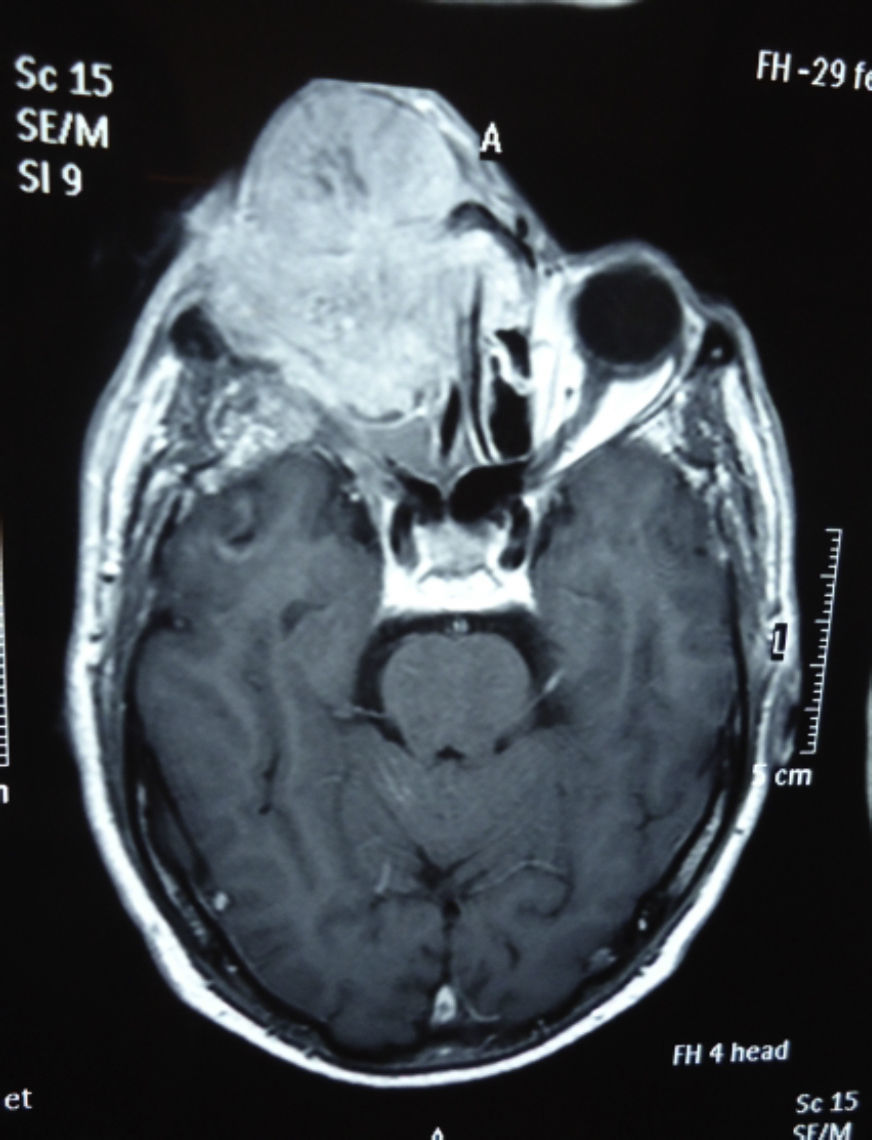
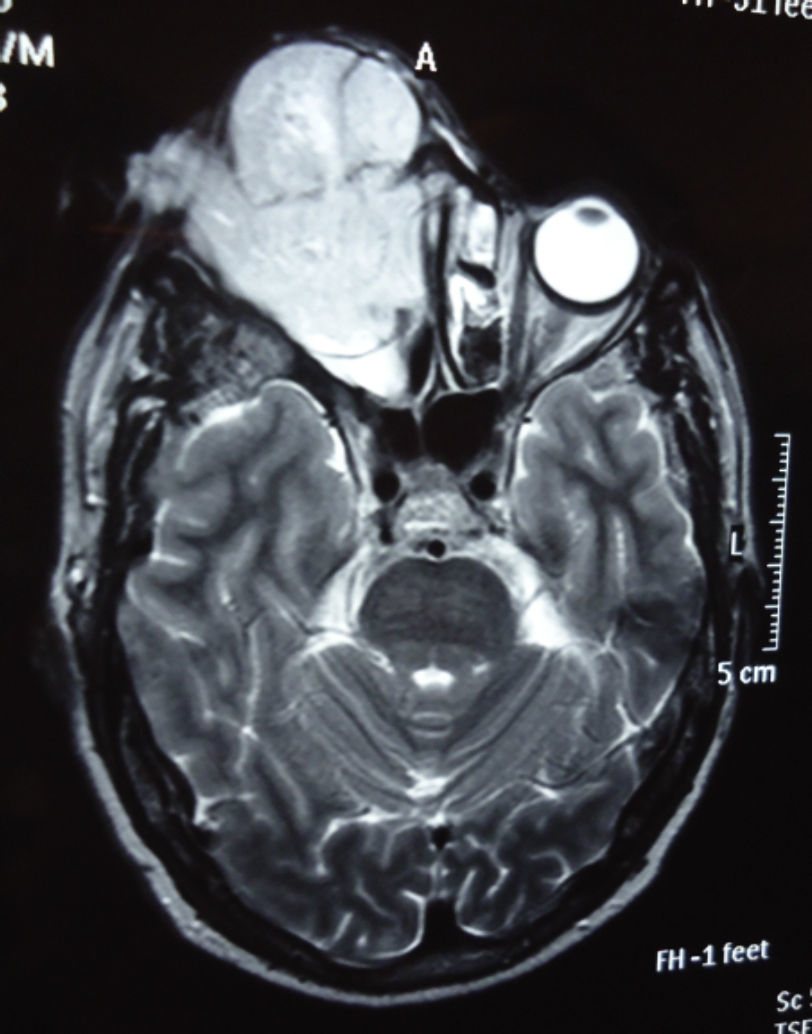
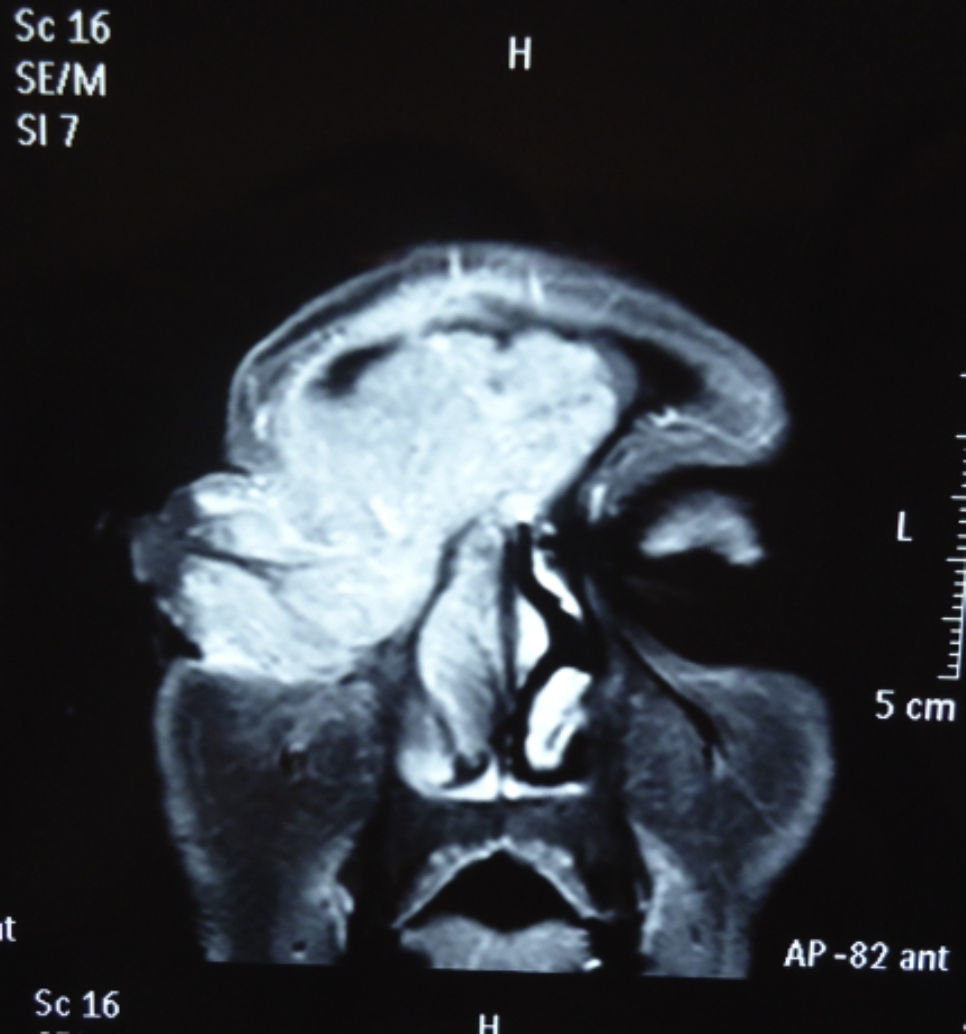
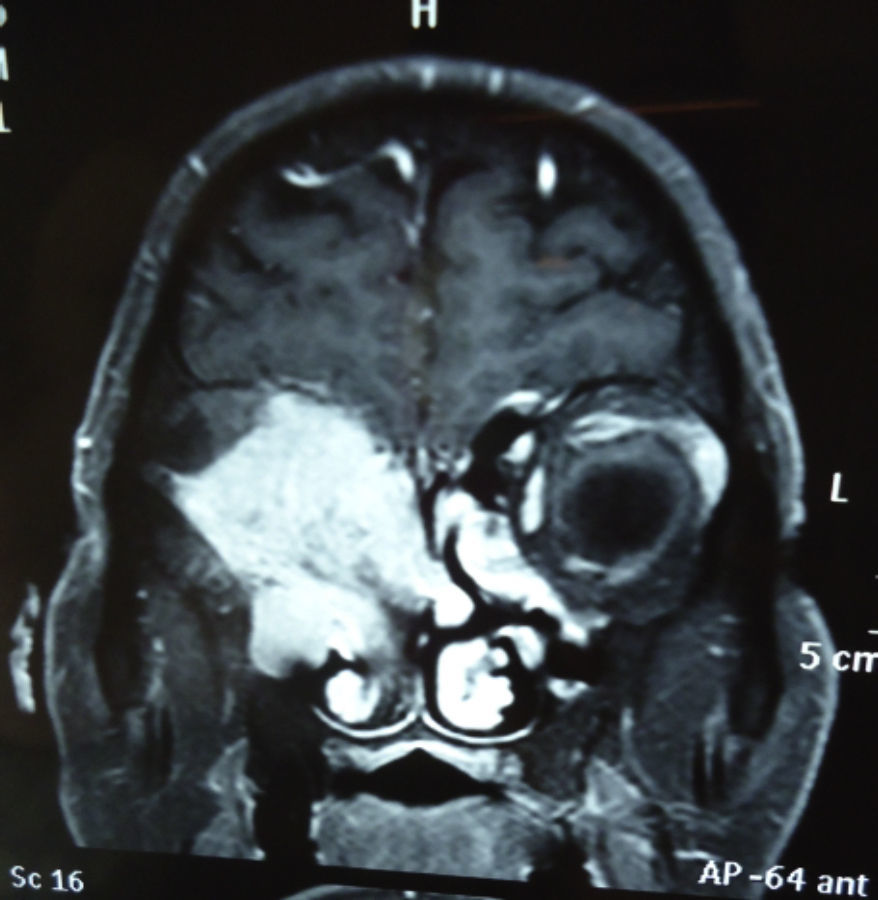
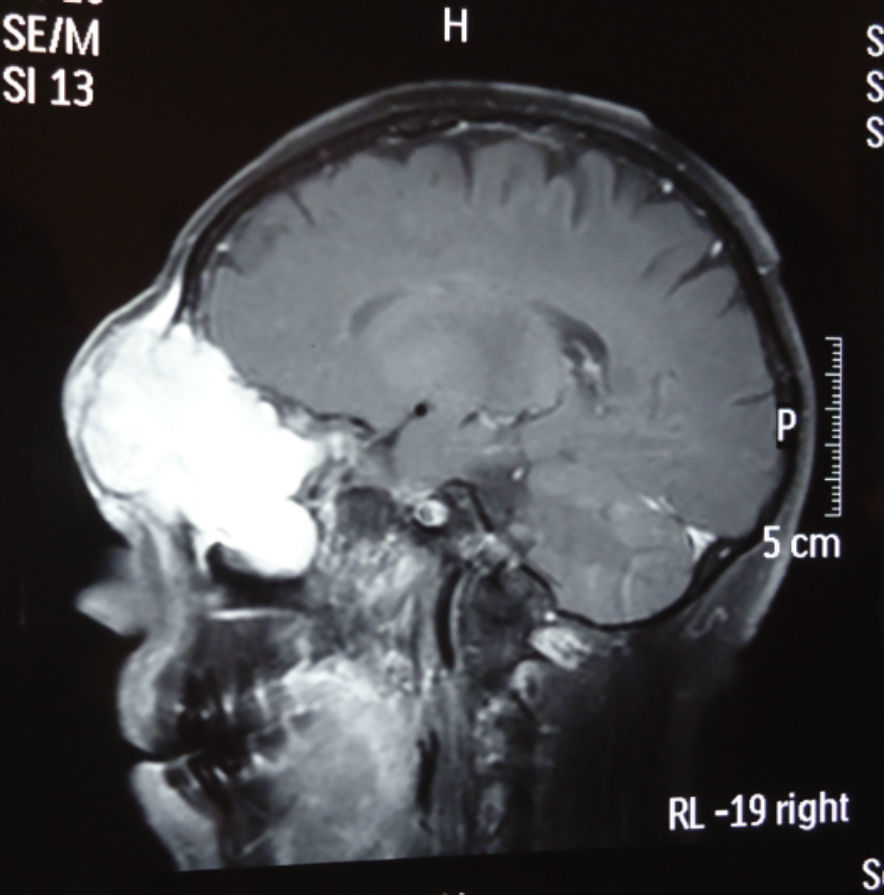
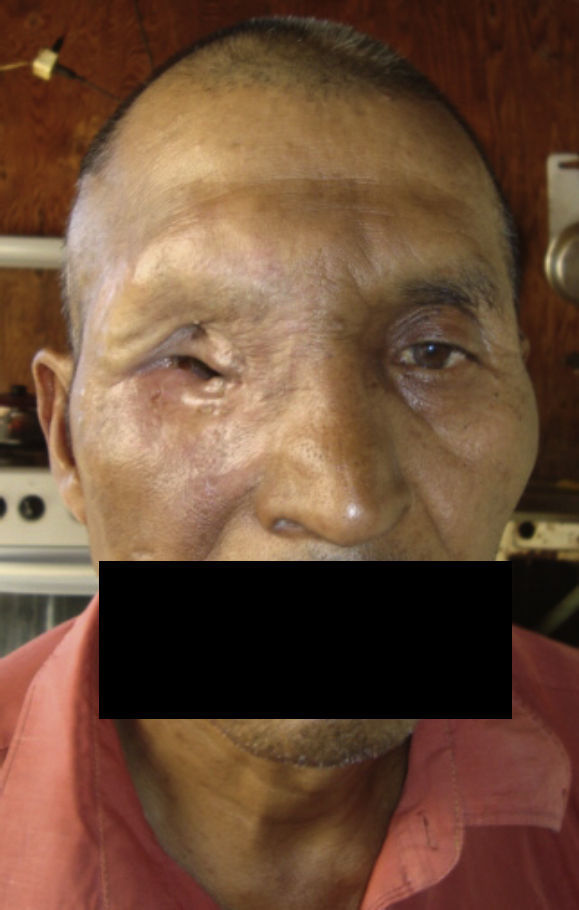
Dos meses después comenzó con un aumento de volumen en región frontal derecha, rápidamente progresivo, abollonado, de consistencia dura, indolora, sin lesión de piel (figs. 7–11). TAC de órbitas evidencia una gran lesión que ocupa la cavidad orbitaria derecha, comprometiendo las celdillas etmoidales y región frontal derecha, con marcada remodelación ósea a nivel del techo y piso orbitario (figs. 12 y 13). Se presenta el caso al comité oncológico, indicando estudio de extensión sistémica mediante punción lumbar: citología de LCR normal; TAC de tórax: 4 nódulos pulmonares derechos inespecíficos, de 5mm; TAC de abdomen y pelvis: normal; cintigrama óseo (Tc99m MDP): aumento de actividad osteoblástica en región periorbitaria derecha, con extensa captación ósea frontal y maxilar, sobrepasando la línea media (fig. 14); y RMN de cerebro (T1, T2, T2 gradiente, FLAIR, difusión/ADC en los planos sagital, coronal y axial, sin y con uso de gadolinio): Voluminoso proceso expansivo en órbita derecha, con extensión intracraneal a nivel bifrontal, en contacto íntimo con parénquima frontobasal a derecha. Invade hueso frontal, pared lateral nasal y pared anterior del antro maxilar ipsilaterales. Con gran componente exofítico a nivel de tejidos blandos adyacentes. Compromete también seno frontal, celdillas etmoidales, fosa nasal, nasofaringe, fosa pterigopalatina y antro maxilar derecho. Intensidad de señal homogénea, morfología lobulada, que refuerza intensamente con medio de contraste. Margen lateral izquierdo de la lesión en contacto con pared orbitaria medial y techo orbitario contralateral, sin invasión a órbita izquierda. Ocupación de celdillas etmoidales izquierdas (figs. 15–20).














Con estos antecedentes se le ingresa en el programa de cuidados paliativos y alivio del dolor, realizando radioterapia externa paliativa: Clinac 600C. Fotones 6 MEV 100cm. de S.S.D. Frontal+órbita+nasal+seno maxilar derecho. AP-LD. 3,000cGy/10 fracciones.
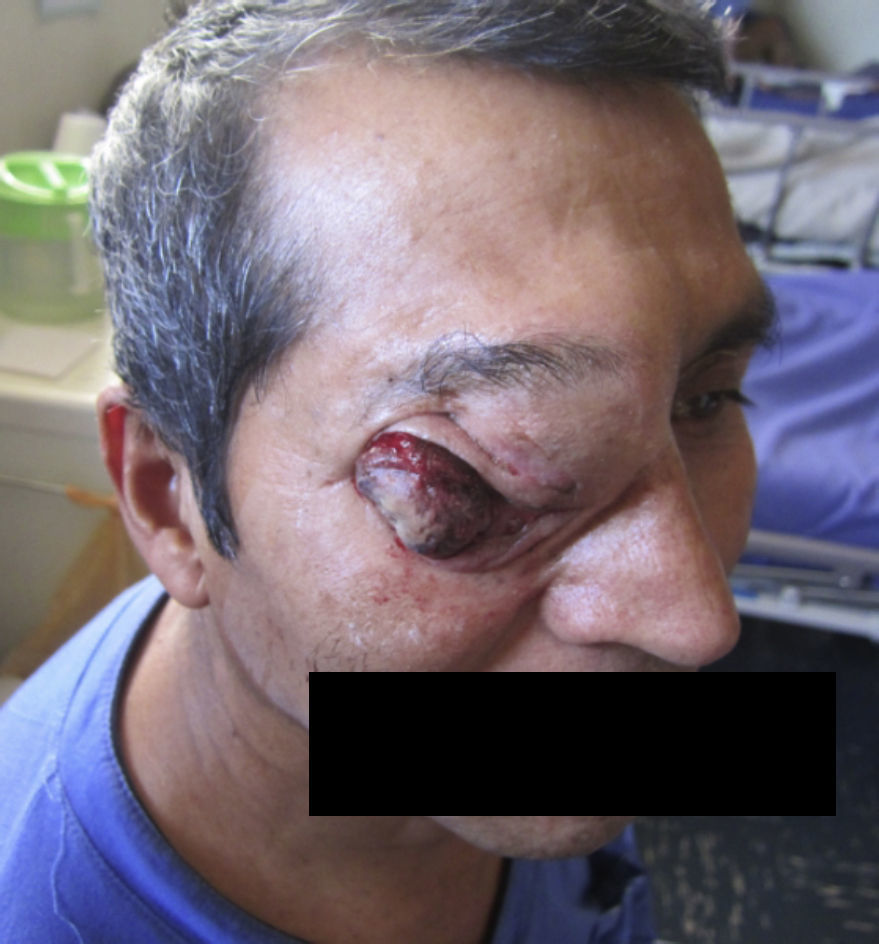
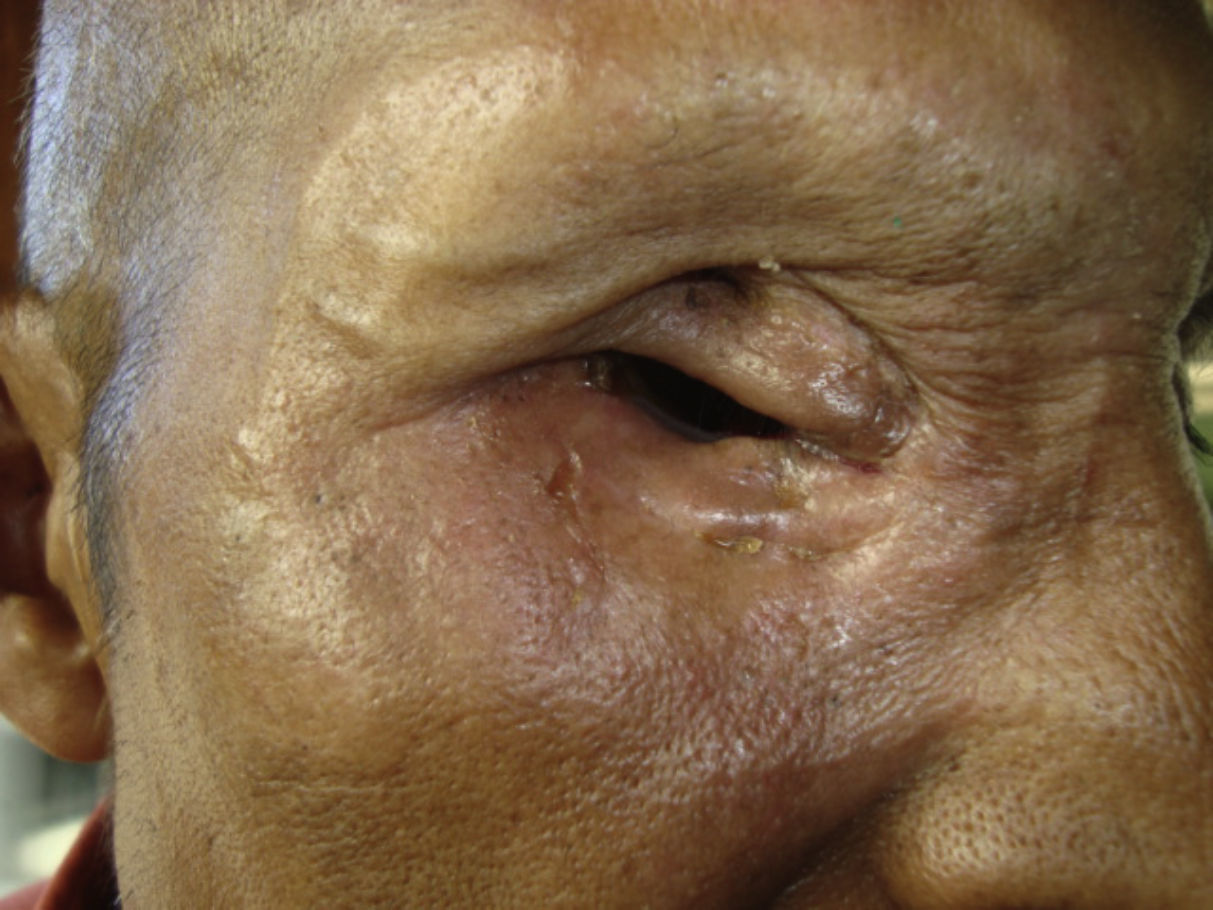
EL paciente evoluciona con importante regresión del tamaño tumoral, disminuye el exudado y se suspende el sangrado (figs. 21 y 22). Dolor EVA 5 al ingreso y EVA 1 al egreso. Actualmente con fármacos analgésicos y controles periódicos.
Discusión

Este tumor fue inicialmente denominado carcinoma primitivo por Badel y Lagrange en 1892, Verhoeff lo nombró teratoneuroma en 1904, y Fuchs lo llamó dictioma en 1908 por la formación de trabéculas de color naranja. Grinker en 1931 utilizó por primera vez el término meduloepitelioma5,7. Zimmerman y Broughton los clasificaron luego en teratoides y no teratoides, y en malignos y benignos3,4,8.
El tipo más frecuente es el no teratoide14, el cual está formado por nidos sólidos o estructuras tubuloglandulares de tamaño variable, muchas de ellas dilatadas y de aspecto quístico. En el interior de los quistes se encuentra un material de características similares al vítreo, el cual se hace más evidente con tinciones para mucopolisacáridos ácidos del tipo azul alciano y hierro coloidal. Dichas formaciones tubulares se encuentran revestidas por un epitelio cilíndrico simple o estratificado, con discreto pleomorfismo celular, hipercromasia nuclear y algunas mitosis. En ocasiones tumores poco diferenciados pueden presentar rosetas de Flexner-Wintersteiner, simulando un retinoblastoma. Estas neoplasias tienen cantidades variables de vasos sanguíneos dispuestos al azar16.
Dos tercios de los meduloepiteliomas son malignos, con una tasa de mortalidad del 10%; la causa de muerte más frecuente es secundaria a extensión intracraneal y recurrencia local5. Los criterios de malignidad son: 1) áreas de células poco diferenciadas semejantes al retinoblastoma, 2) presencia de componente sarcomatoide o anaplásico, 3) invasión a la úvea o esclerótica, 4) pleomorfismo celular y 5) mitosis3,4,8,11.
Por inmunohistoquímica las células del meduloepitelioma presentan inmunorreactividad para sinaptofisina, proteína S-100, NSE, vimentina; y por su naturaleza pluripotencial y polifenotípica pueden expresar GFAP, neurofilamento, diversas citoqueratinas, desmina y HMB-459. Las células neoplásicas sintetizan una sustancia rica en ácido hialurónico semejante al humor vítreo, que es positiva a la tinción de azul alciano y negativa al PAS4,15.
Por microscopia electrónica, Wakakura y Lee identificaron 3 tipos celulares: epitelial, neuronal e intermedio. Las células neuronales se disponen en estructuras rosetoides, las células epiteliales en trabéculas, rodeando los espacios quísticos con pigmento melánico maduro e inmaduro, y las células intermedias tienen características de ambas15.
Desde el punto de vista genético no se ha identificado una alteración constante en el meduloepitelioma10,12. Betts et al.17 han informado translocación t1,16, deleción (6q) y monosomía 15, pero estos datos fueron aislados y no concluyentes.
El epitelio ciliar adulto tiene una población de células progenitoras neurales en condiciones no proliferativas18,19. Coles et al. identificaron células similares en el iris, pars plicata y pars plana del ojo humano, que en cultivos de tejido desarrollan esferas de células indiferenciadas pigmentadas y no pigmentadas19. La densidad de estas células progenitoras es mayor en la pars plicata, y fueron halladas en estadios de desarrollo posnatal hasta la sexta década, mostrando su mantención desde el nacimiento hasta edades avanzadas. De esta forma, se puede considerar que el meduloepitelioma podría desarrollar células neuroepiteliales inmaduras en ojos bien desarrollados. En algunos estudios, la inyección intraocular de células madre embrionarias en ratones «nude» desarrolla meduloepiteliomas20. Carrillo y Streeten mostraron además que la aparición tardía en adultos de estas neoplasias podría deberse a un rápido crecimiento luego de la transformación neoplásica de un meduloepitelioma benigno no detectado previamente21. Así, el desarrollo de un meduloepitelioma en el adulto podría deberse a: 1) bajo índice de crecimiento del tumor en casos de neoplasias benignas, 2) transformación maligna de una neoplasia benigna previamente no identificada, y 3) desarrollo de una nueva neoplasia a partir de células progenitoras del epitelio ciliar.
El diagnóstico debe sospecharse por los datos clínicos, complementados por estudios de imagen (ultrasonografía, TAC, RMN), pero el diagnóstico definitivo es histopatológico1,3,4,10,20,22–24.
Ecográficamente, en el modo B se presenta como una tumoración en forma de domo, densidad heterogénea, con grandes espacios quísticos en su interior, localizada en la región del cuerpo ciliar. Las formas de meduloepitelioma que contienen cartílago producen ecos de extremada alta reflectividad. Se pueden desprender células hacia el vítreo, desarrollando quistes a ese nivel. En el estudio A estandarizado la reflectividad es alta, estructura interna irregular, con moderada vascularidad y múltiples espacios quísticos en su interior. Estas características se explican por la heterogeneidad de la arquitectura histológica de la lesión16.
Este tumor en general está confinado a la órbita, y rara vez produce invasión local y metástasis4,25. El tratamiento es controvertido. Las lesiones pequeñas y circunscritas han sido tratadas con resección local, con alto riesgo de recurrencia; en las lesiones poco definidas e invasivas el tratamiento de elección es la enucleación. Los casos limitados al ojo tienen buen pronóstico, debido a que presentan crecimiento lento, y las metástasis hematógenas y linfógenas son raras, con alrededor de un 8%, especialmente a parótida, ganglios linfáticos cervicales y submandibulares25,26, encontrándose en todas ellas extensión extraescleral del tumor14. De esta forma, los casos mal delimitados o de diagnóstico tardío tienen pronóstico malo, porque son altamente recurrentes y localmente agresivos3,4,8,10,18–20.
El diagnóstico diferencial se debe realizar con el rabdomiosarcoma, retinoblastoma, neuroblastoma, malformaciones vasculares, quistes hemáticos, glioneuroma y con la hiperplasia primaria del vítreo5,9,12,20. En los casos pigmentados, el diagnóstico diferencial se hace con lesiones melanocíticas (melanoma, melanocitoma, nevos), tumor neuroectodérmico pigmentado de la infancia, quistes iridociliares pigmentados, adenomas o adenocarcinoma del epitelio pigmentado4,10,11.
ConclusionesEl meduloepitelioma es un tumor orbitario de diagnóstico excepcional en el adulto debido a su extremada baja frecuencia. El bajo número de casos descritos en la literatura médica hacen difícil caracterizar su forma de presentación, evolución y manejo. Su comportamiento aún sigue siendo incierto, aunque al parecer se trata de un tumor con escasas metástasis hematógenas y linfógenas, aunque muy agresivo a nivel local, con recurrencias frecuentes, necesitándose un manejo bajo observación cuidadosa y terapéutica agresiva.
FinanciamientoLos autores no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
