


Las ictiosis constituyen un grupo de padecimientos hereditarios con alteraciones en la queratinización. La segunda forma más frecuente es la ligada a X con una prevalencia de 1:2,000–6,000 varones. En esta variedad se observan lesiones corneales características.
Caso clínicoDos hermanos de 19 y 18 años que referían mala agudeza visual desde la infancia la cual mejoraba con el uso de corrección refractiva. A la exploración con lámpara de hendidura se encontraron opacidades blanco-grisáceas puntiformes y granulares en estroma posterior de la córnea de ambos ojos de los 2 pacientes. Se utilizó microscopia confocal, la cual demostró lesiones hiperdensas pre-Descemet Sistémicamente se observaron lesiones dérmicas a manera de escamas, hiperqueratósicas e hiperpigmentadas.
ConclusionesLos datos clínicos fueron compatibles con ictiosis ligada a X. En este padecimiento están descritas opacidades pre-Descemet en el 25-50% de los casos las cuales no interfieren con la visión pero pueden ocasionar úlceras corneales recurrentes.
The ichthyoses comprise a group of heterogeneous hereditary diseases characterized by altered keratinization. The X-linked recessive form is the second most common with prevalence of 1:2,000–6,000 men. Corneal involvement has been described in these patients.
Case reportTwo brothers aged 19 and 18 years old referring blurred vision since childhood that improved with refractive correction. Slit lamp exam revealed multiple white-grey flourlike opacities in the posterior corneal stroma in both patients. Confocal microscopy showed hyperreflectic opacities at the level of the posterior stroma. Physical exam revealed dermal scale like hyperpigmented, hyperkeratosic lesions in both patients.
ConclusionsClinical features and family pedigree were consistent with X-linked ichthyosis. In this disease pre-Descemet opacities that do not interfere with vision but may cause recurrent corneal ulcers have been found in 25 to 50% of patients.
Las ictiosis comprenden un grupo heterogéneo de enfermedades hereditarias que se caracterizan por alteraciones en la queratinización con la consecuente formación de escamas. Se han descrito diversas formas las cuales se clasifican según el patrón de herencia, las manifestaciones clínicas así como por el defecto bioquímico. La forma ligada a X es la segunda más común con una prevalencia de 1:2,000-6,000 varones. Las manifestaciones clínicas aparecen durante el primer año de vida y consisten en escamas grandes, gruesas, adherentes e hiperpigmentadas, principalmente en regiones extensoras de las extremidades, región lateral de la cara, cuello y cuero cabelludo1. El defecto bioquímico es la deficiencia de sulfatasa esteroidea con aumento secundario del sulfato de colesterol sérico y el depósito del mismo en el estrato córneo de la dermis. Por otro lado, en estos pacientes se han descrito opacidades corneales, como puntos de color blanco-grisáceo en el estroma posterior; estas lesiones no interfieren con la visión2.
Casos clínicosSe presentaron al servicio de consulta externa del Hospital «Dr. Luis Sánchez Bulnes» de la Asociación para Evitar la Ceguera en México 2 hermanos del sexo masculino de 19 años (caso 1) y 18 años (caso 2); referían mala agudeza visual desde la infancia que mejoraba con el uso de corrección refractiva.
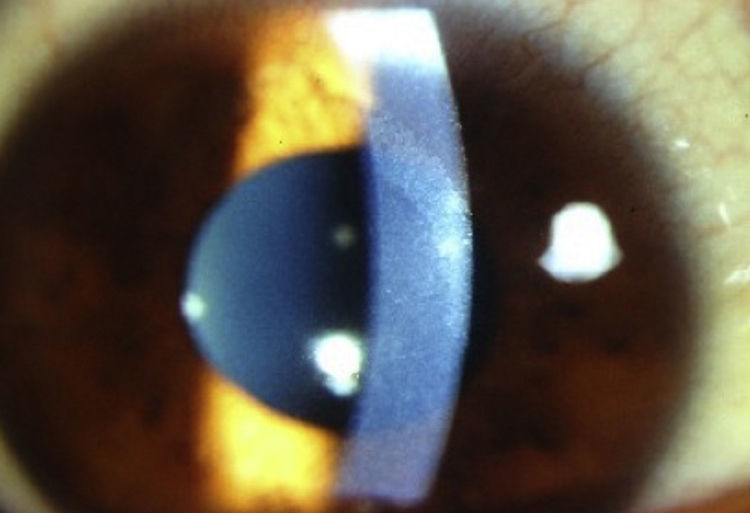
El caso 1 refería antecedente de crisis convulsivas en tratamiento con valproato. A la exploración oftalmológica se encontró agudeza visual en ambos ojos (AO) de 20/80; agudeza visual mejor corregida de 20/25 en ojo derecho y 20/20 en ojo izquierdo, presión intraocular de 15mmHg en AO. En la biomicroscopia con lámpara de hendidura se encontraron múltiples opacidades corneales blanco-grisáceas, puntiformes y granulares en estroma corneal posterior (fig. 1), el resto de la exploración no mostró alteraciones.
, caso 1.")
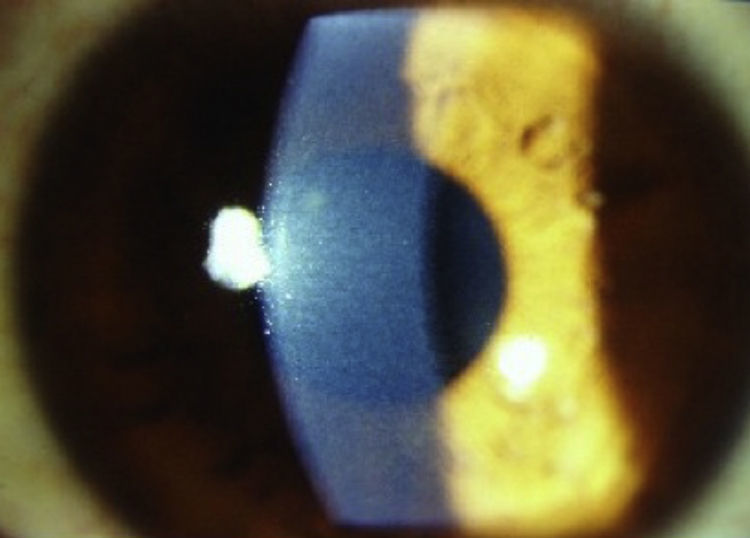
El caso 2 tenía una agudeza visual de 20/50 y agudeza visual mejor corregida de 20/25 en AO y presión intraocular de 16mmHg en AO. En el examen con lámpara de hendidura se observaban opacidades corneales similares a las descritas en el caso anterior (fig. 2), sin evidencia clínica de otras alteraciones oftalmológicas.
, caso 2.")
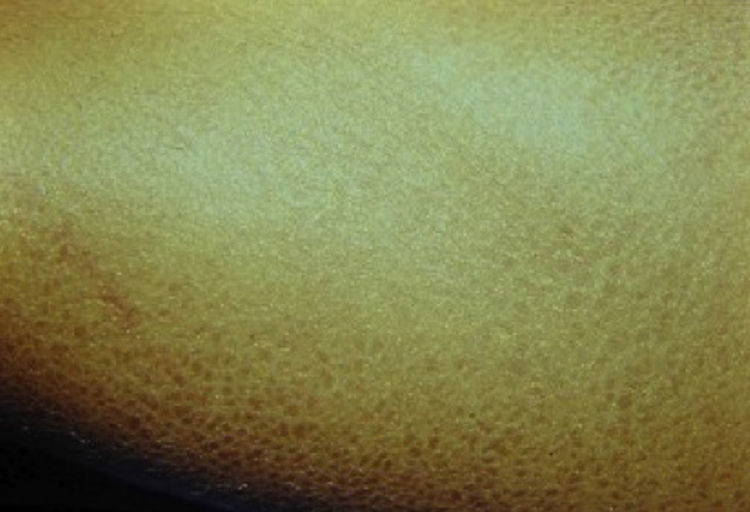
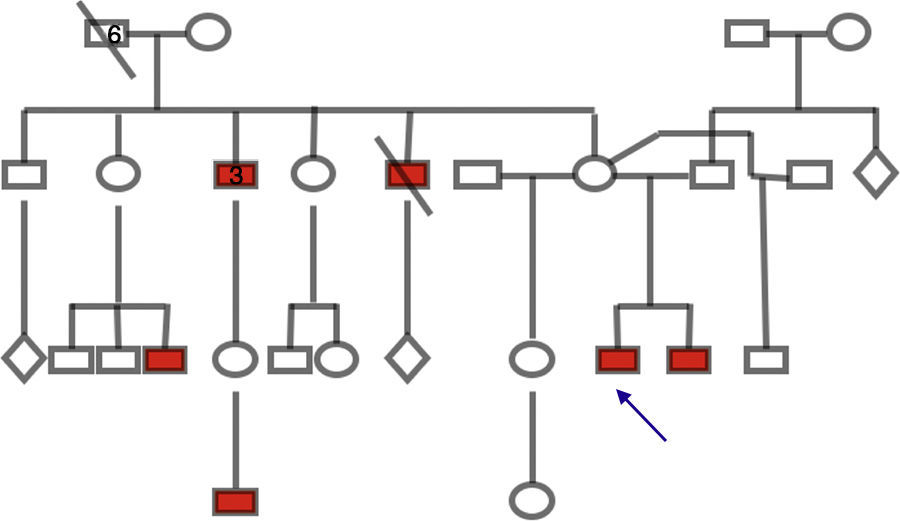
En la exploración sistémica de ambos pacientes se encontraron lesiones dérmicas con aspecto escamoso, hiperpigmentadas e hiperqueratinizadas en las extremidades inferiores, el dorso de los pies y el abdomen (fig. 3). El árbol genealógico demostró varios miembros del sexo masculino con lesiones dérmicas similares, por lo que se estableció un patrón de herencia ligada a X recesiva (fig. 4).

, compatible con herencia ligada a X. La flecha muestra el caso índice.")
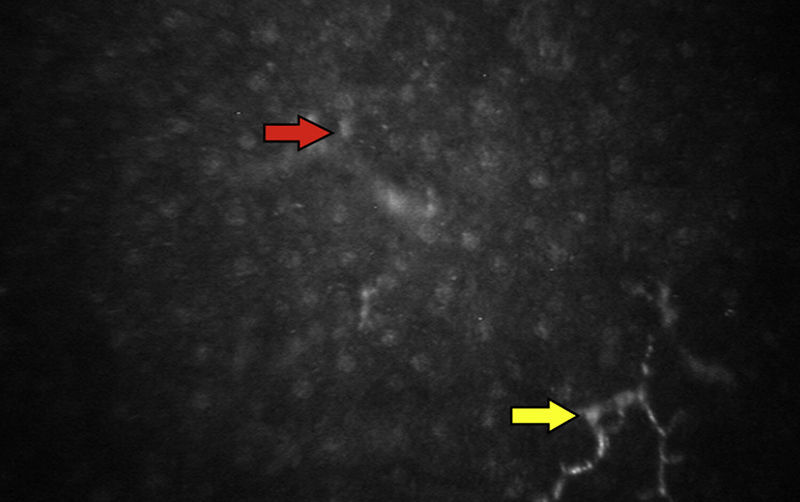
En los 2 casos se realizó estudio de microscopia confocal la cual reportó opacidades hiperreflectantes a nivel del estroma corneal posterior (fig. 5).
Discusión; así como fibras nerviosas de disposición vertical con morfología alterada (flecha amarilla).")
Las ictiosis comprenden un grupo de enfermedades hereditarias con alteraciones en la queratinización, formándose en la piel escamas, de aquí el término de ictiosis proveniente del griego y que significa «pescado»1. En la variedad ligada a X se ha demostrado deficiencia de la enzima sulfatasa esteroidea; esta anormalidad es específica y no se presenta en otras variedades de ictiosis3. La determinación sérica de la sulfatasa esteroidea no es necesaria para el diagnóstico del padecimiento cuando se encuentran las características clínicas y los datos familiares compatibles con el diagnóstico, como en el presente caso.
Las lesiones corneales descritas en el padecimiento consisten en opacidades blanco-grisáceas localizadas en el estroma posterior. Se identifican mediante el examen con lámpara de hendidura y solo se han descrito en esta variedad de ictiosis. Las mujeres portadoras pueden llegar a mostrar estas opacidades usualmente después de la adolescencia. Con el uso de la microscopia electrónica se han descrito las alteraciones histológicas, que consisten en colecciones subepiteliales de material proteináceo con gránulos electrodensos, presencia de espacios vacíos entre y dentro de las lamelas de colágena y reorganización de las mismas4.
Es importante señalar que las alteraciones corneales no interfieren en la visión de los pacientes y por su aspecto pueden llegar a confundirse con una distrofia corneal por lo que es importante la revisión sistémica de los pacientes ante estos hallazgos y de esta manera identificar las características de la piel2. Dichas lesiones corneales pueden predisponer a la aparición de úlceras corneales recurrentes. Existen reportes previos del uso de la microscopia confocal para la detección de depósitos corneales5.
Entre las manifestaciones sistémicas de la ictiosis ligada a X se describe criptorquidia en el 10-20% de los casos y aumento en el riesgo de cáncer testicular no relacionado con la falta de descenso testicular; también se describe déficit de atención, trastornos de hiperactividad, epilepsia y malformaciones intracraneales. Se ha considerado que estas alteraciones asociadas se explican por un síndrome de genes contiguos, en ciertos casos asociados a microdeleciones cromosómicas3,6. En el presente reporte encontramos historia de crisis convulsivas en uno de los pacientes, sin embargo no se realizó estudio genético del caso.
ConclusionesLas características clínicas y el árbol genealógico fueron compatibles con el diagnóstico de ictiosis ligada a X. En este padecimiento están descritas opacidades pre-Descemet en el 25-50% de los casos las cuales no interfieren con la visión pero pueden propiciar la aparición de úlceras corneales recurrentes.
El caso es interesante por la diferenciación con distrofias corneales estromales, y no encontramos en la literatura otro reporte de los hallazgos de la microscopia confocal de estas alteraciones.
Para el oftalmólogo es importante considerar la evaluación sistémica de algunos casos con el fin de establecer diagnósticos integrales certeros.
FinanciamientoLos autores no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
