


El osteoma coroideo es un tumor intraocular benigno, raro, compuesto de hueso calcificado maduro, que se presenta de manera típica en mujeres adultas jóvenes.1,2 Los hallazgos a largo plazo de este tumor benigno incluyen crecimiento del tumor en 51%, descalcificación del tumor en 46%, desarrollo de neovascularización en 31%, y una agudeza visual de 20 ¿200 o mayor en 56% de los pacientes.3 Tiene un origen coristomatoso y existen factores hormonales y metabólicos en su etiología. El tratamiento se basa en la observación de los casos asintomáticos sin compromiso visual importante o complicaciones. Las opciones terapéuticas actuales incluyen fotocoagulación láser, terapia fotodinámica y agentes antiangiogénicos.
El síndrome de Crouzon es una anomalía rara, hereditaria con carácter autosómico dominante, causado por cierre prematuro de las suturas craneales (craneosinostosis).
La histiocitosis de células de Langerhans abarca un espectro de trastornos no neoplásicos con manifestaciones variables. El dato común es la proliferación de las células de Langerhans, un tipo de macrófagos tisulares que, entre otras características, contienen gránulos de Birbeck. Es rara, pero afecta la órbita y puede comenzar en ella. No se conoce la existencia de una asociación entre estas patologías.
En este informe presentamos el caso poco frecuente de un paciente de nueve años con síndrome de Crouzon, antecedente de histiocitosis de células de Langerhans y osteoma coroideo.
¿ Informe del caso
Niño de nueve años de edad, originario y residente de Veracruz, enviado a valoración oftalmológica por exoftalmos de ambos ojos. Madre de 25 años, analfabeta, aparentemente sana. Padre de 28 años de edad, con primaria incompleta, tabaquismo y alcoholismo ocasional; antecedente de tuberculosis pulmonar; dos hermanos de diez y seis años de edad, aparentemente sanos.
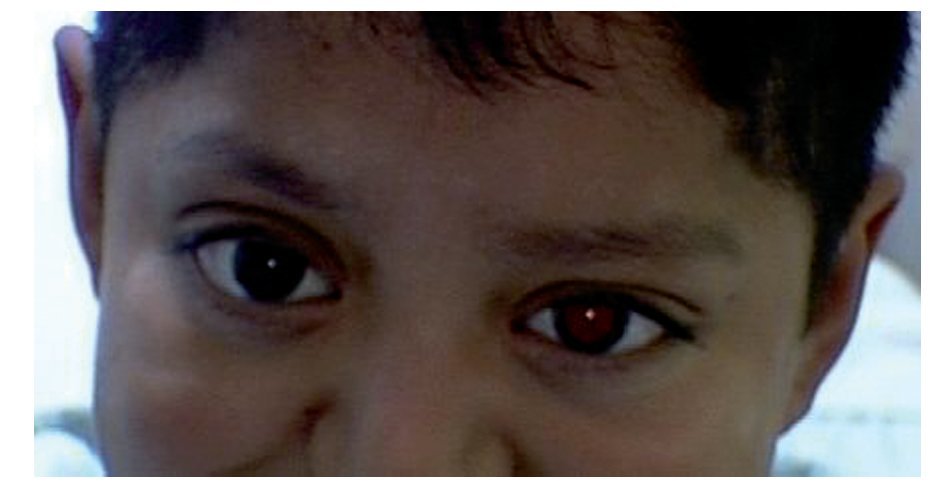
El paciente fue producto de la segunda gestación, con embarazo de término, obtenido por vía vaginal, eutócico, con peso al nacer de 3000 g y talla de 48 cm. Fue diagnosticado al nacer como síndrome de Crouzon. A los dos años de edad se le realizó el diagnóstico de histiocitosis de células de Langerhans, tratado con quimioterapia, y actualmente se encuentra en vigilancia. Se le practicó cirugía de cuello no especificada, cirugía de cráneo por craneosinostosis (remodelación ósea), recibiendo transfusión sanguínea (Figura 1).
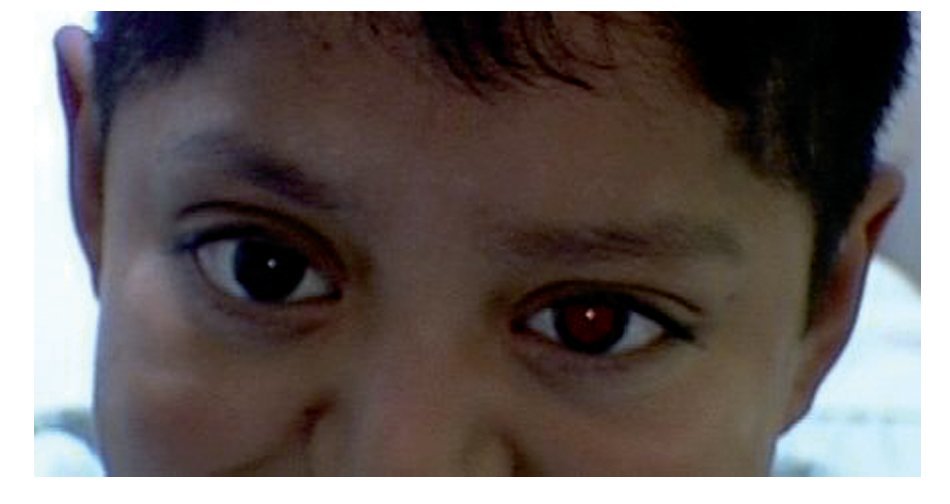
¿ Figura 1. Aspecto clínico del paciente; niño de nueve años de edad.
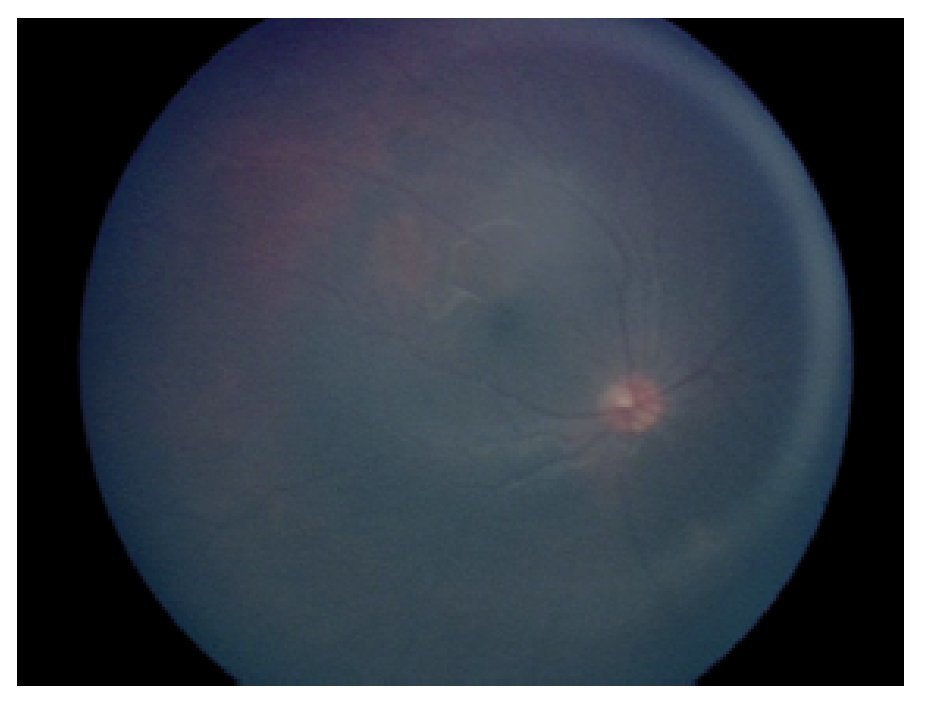
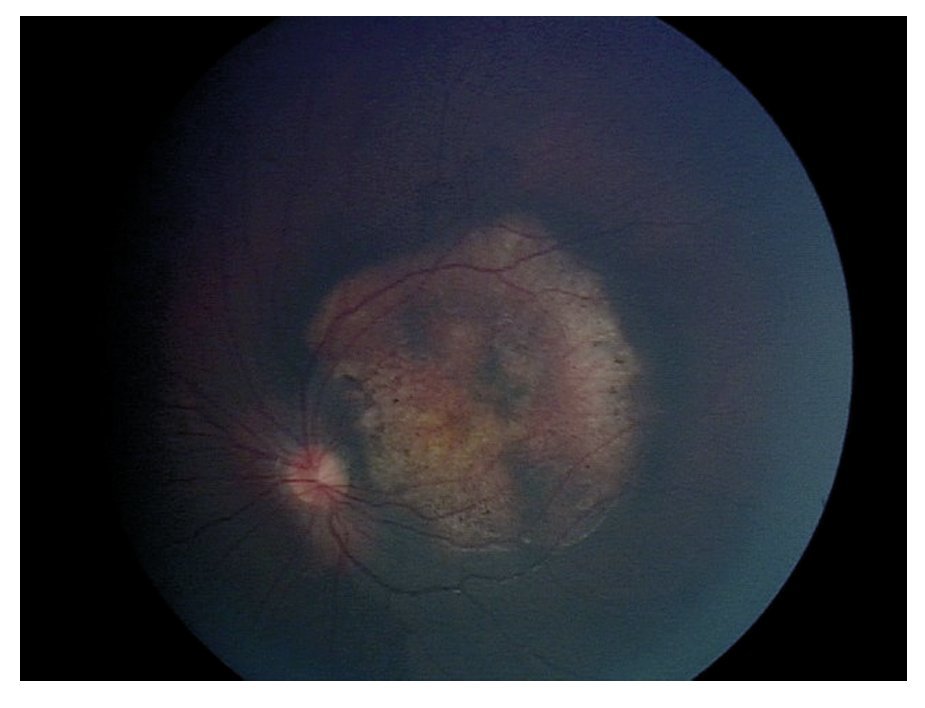
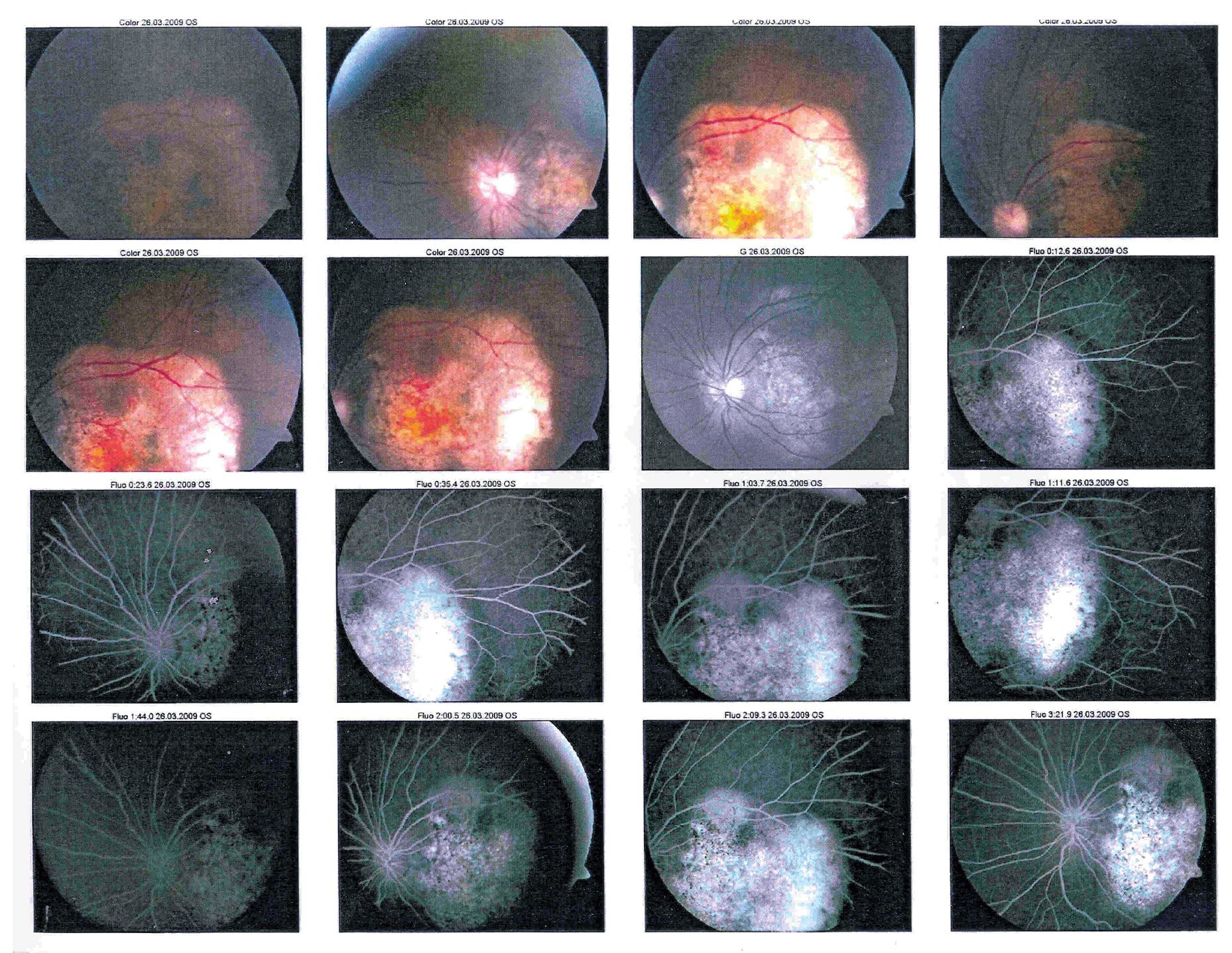
En la exploración oftalmológica se encontró agudeza visual de ojo derecho 20/40 y ojo izquierdo 20/30. Presentó exoftalmos bilateral, mayor de ojo izquierdo, pero con cierre palpebral adecuado en ambos ojos. La refracción del ojo derecho -1.25 » -1.00 x 60, y del ojo izquierdo - 0.75» -0.75 x 10. No se encontró alteración a la biomicroscopía. Exploración de fondo de ojo con oftalmoscopía indirecta bajo midriasis farmacológica (fenilefrina y tropicamida) en ojo derecho sin alteraciones aparentes, en ojo izquierdo se observó lesión yuxtapapilar blanco amarillenta, plana, de aproximadamente 20 diámetros papilares con involucro del área macular (Figura 2 y 3).
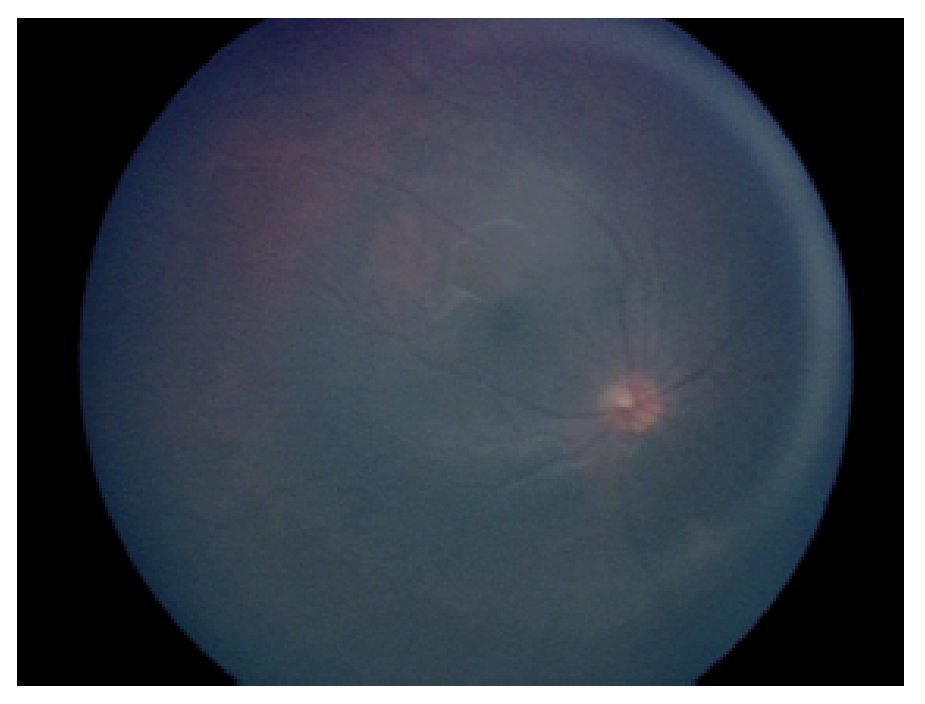
¿ Figura 2. Fotografía de fondo de ojo derecho.
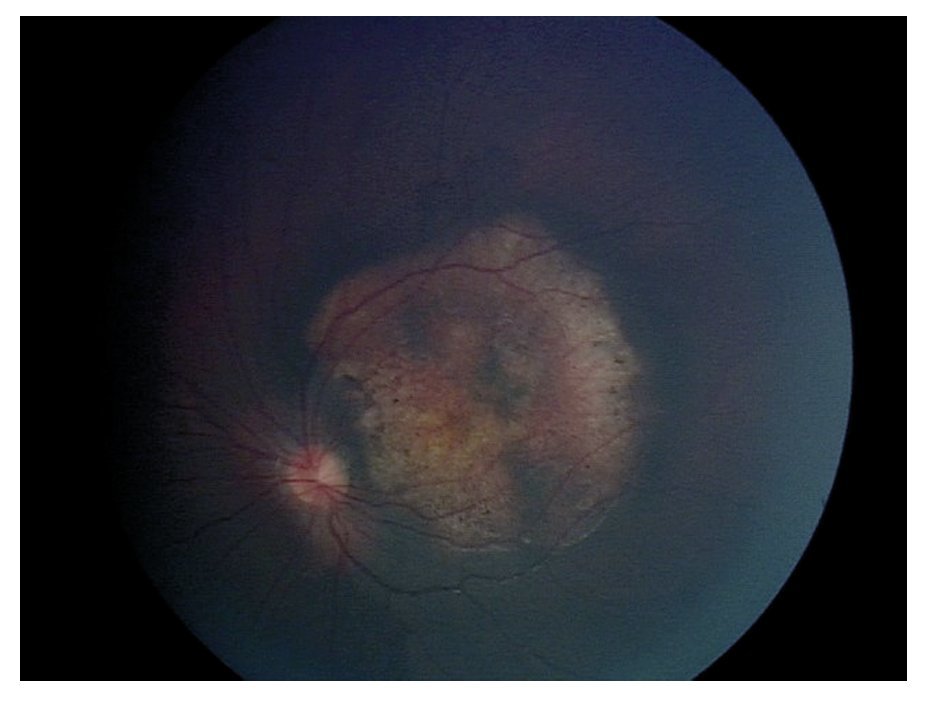
¿ Figura 3. Fotografía del fondo de ojo izquierdo.
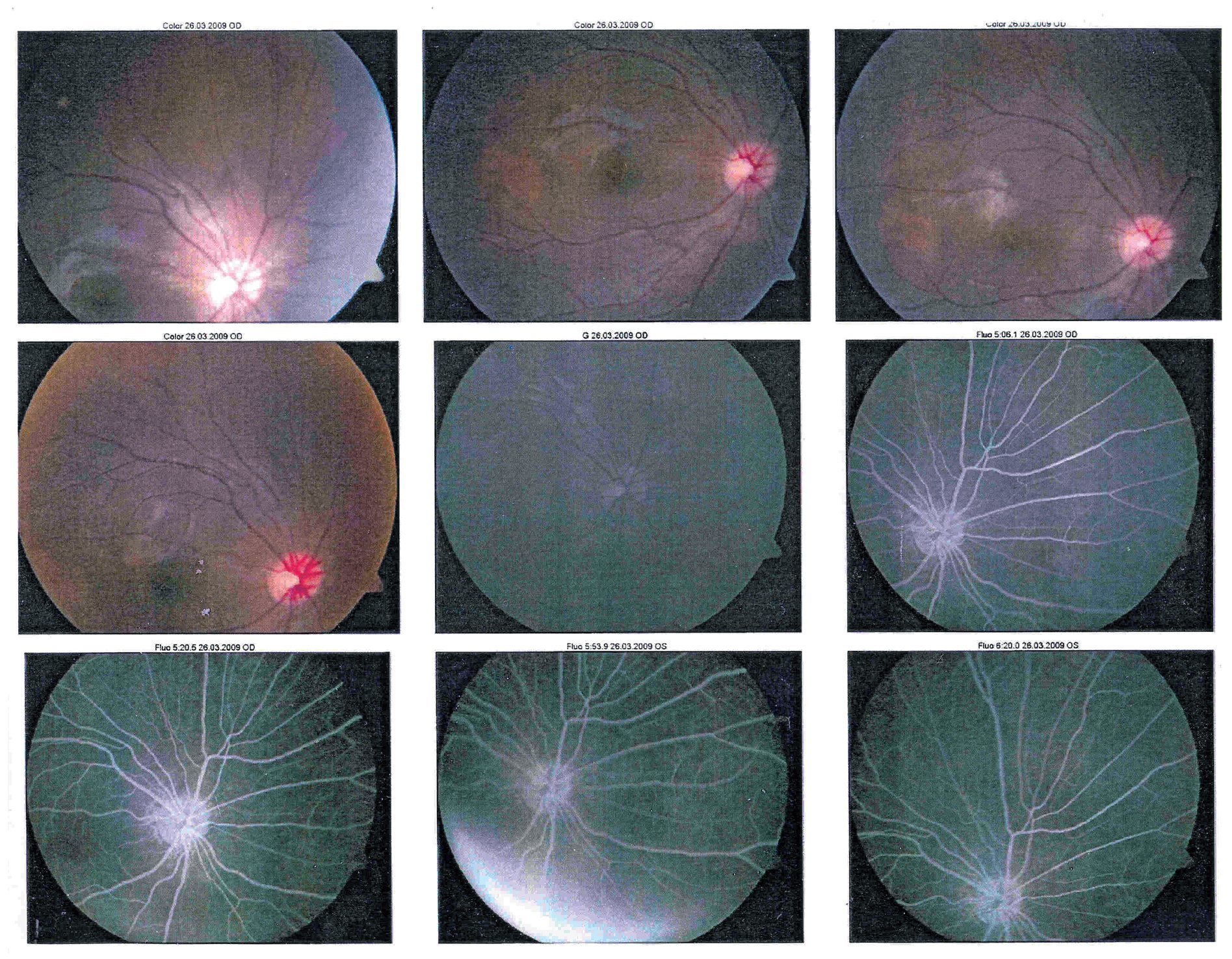
En la fluorangiografía (FAG) de ojo izquierdo se demostró hiperfluorescencia de la lesión en fases tempranas, que aumentaba de intensidad pero no de tamaño (Figura 4 y 5).
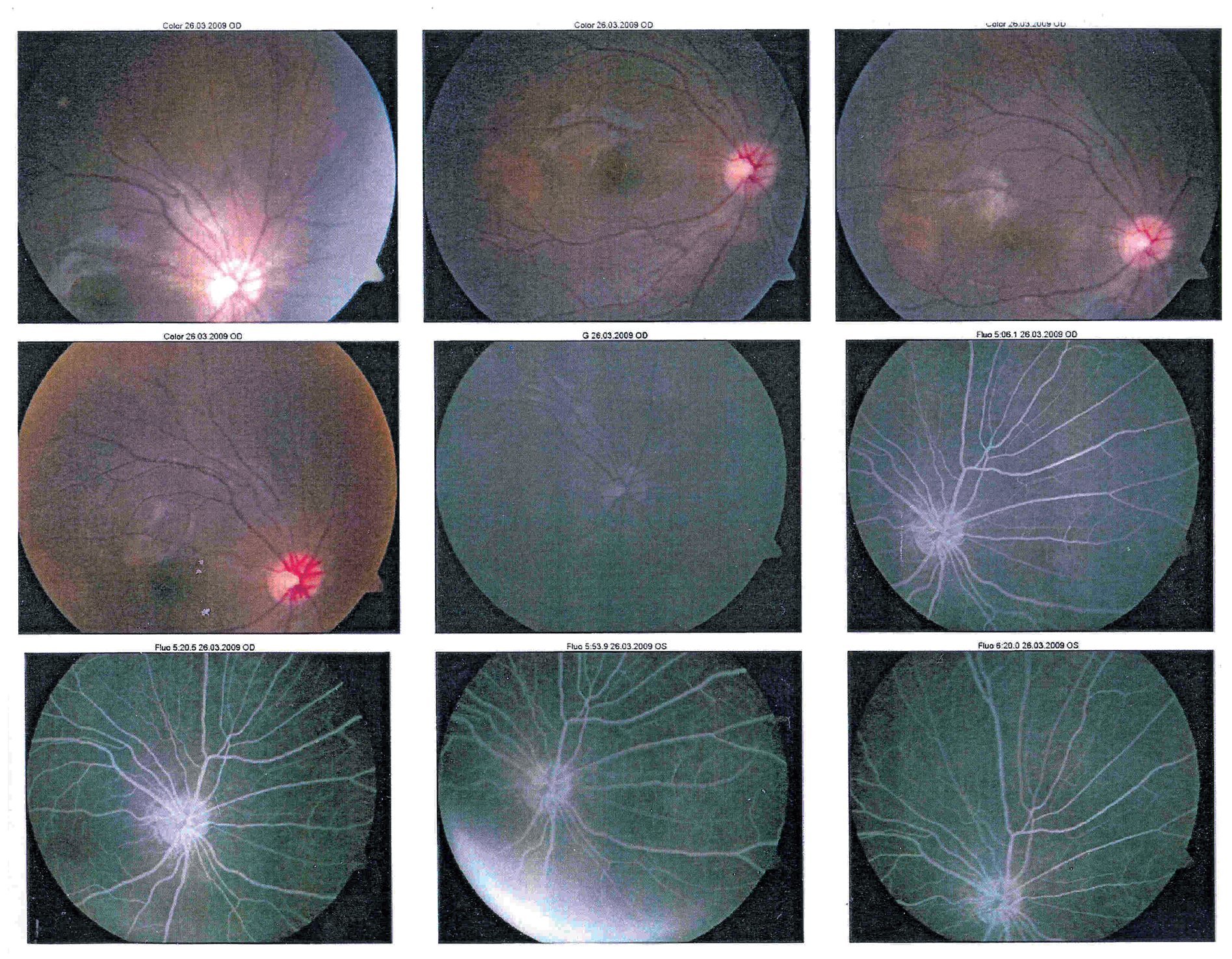
¿ Figura 4. Fluorangiografía del ojo derecho.
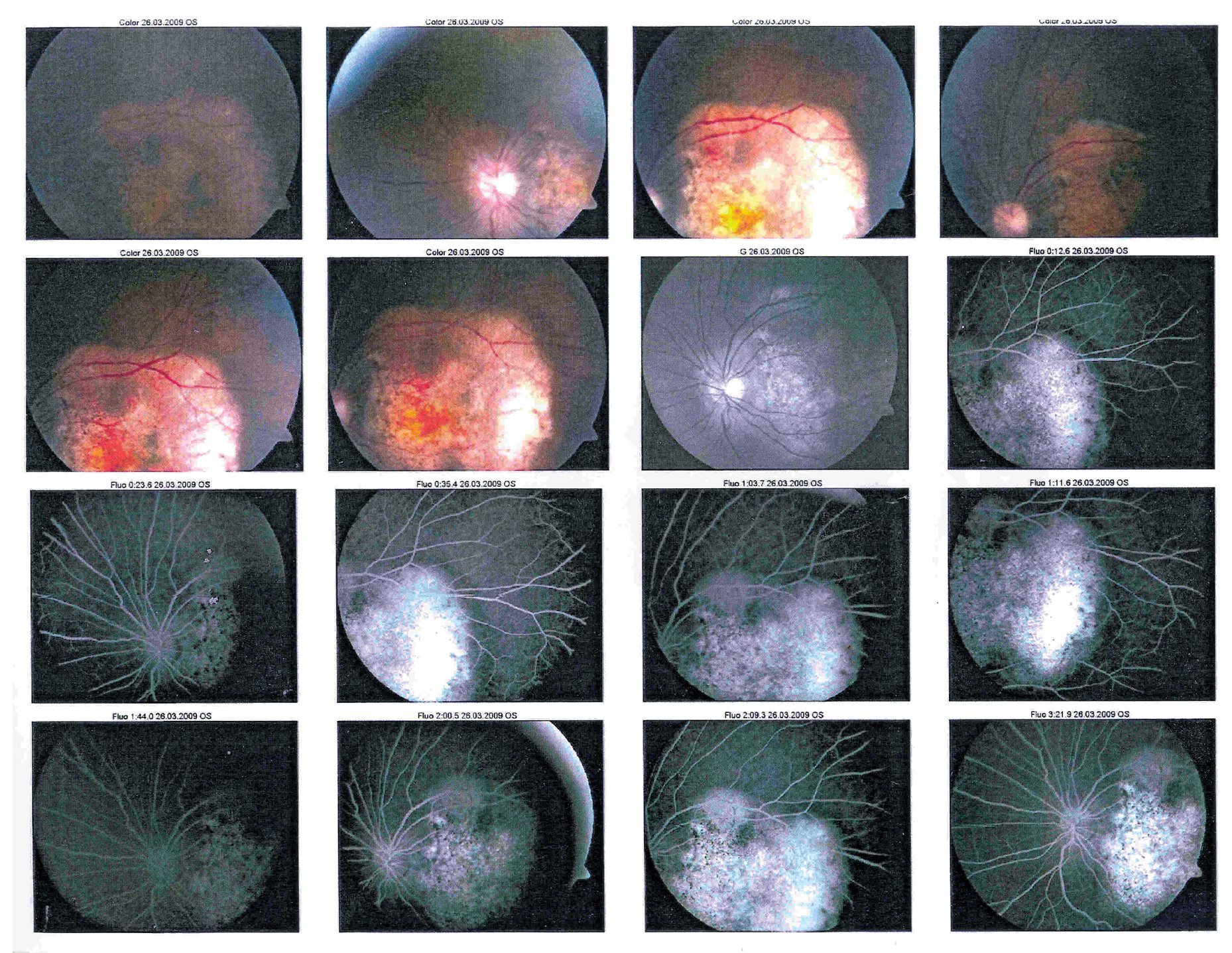
¿ Figura 5. Fluorangiografía del ojo izquierdo.
La ultrasonografía (US) mostró una imagen con densidad cálcica en la pared posterior de la retina del globo ocular izquierdo adyacente a la emergencia del nervio óptico, con diámetro aproximado de 8 mm.
En la tomografía axial computada se encuentran cambios quirúrgicos por remodelación ósea y evidencia de exoftalmos bilateral por el antecedente de síndrome de Crouzon, así como imagen de calcificación a nivel del polo posterior del globo ocular izquierdo.
Se realizó electroencefalograma (solicitado por neurocirugía) que refiere lentificación generalizada, asimétrica, por menor voltaje en el hemisferio cerebral izquierdo, sin actividad paroxística.
¿ Discusión
En 1976, Reese publicó el primer caso de osteoma coroideo, pero fue erroneamente diagnosticado como hemangioma coroideo osificante. El término "osteoma coroideo" fue introducido por Gass en 1978, quien describió los casos de cuatro mujeres jóvenes sanas con hallazgos oftalmológicos característicos. Otro término para esta condición fue "coristoma oseo".4
Existen pocos casos bien documentados que ocurren después de inflamación ocular. Se han publicado casos familiares con involucro multigeneracional, pero la mayoría de los casos aparecen esporádicamente.5,6 El papel de factores hormonal y metabólico en la etiología del osteoma coroideo sigue en especulación.
En nuestro paciente consideramos que el síndrome de Crouzon, la histiocitosis de Langerhans y el osteoma coroideo en el ojo izquierdo corresponden a entidades separadas, sin ser las primeras una causa de la patología ocular.
El osteoma coroideo comúnmente se presenta como una lesión solitaria localizada en la región yuxtapapilar de un ojo en una mujer joven sana. Es raro en la infancia temprana y en adultos mayores.4 La edad de nuestro paciente no es frecuente para la presentación de la patología, así como ser del sexo masculino.Los síntomas más comunes son disminución visual, metamorfopsias y defectos del campo visual. La mayoría de los pacientes son asintomáticos. La neovascularizacion coroidea es la principal complicación del osteoma coroideo y ocurre en 50% de los casos en un lapso de 10 años.7
Dentro de las herramientas diagnósticas tenemos, aparte de la apariencia oftalmológica, la fluorangiografía, la angiografía con indocianina, la tomografía óptica coherente, la ultrasonografía, la tomografía computada y la imagen de resonancia magnética.4 Nuestro paciente cuenta con fluorangiografía y la ultrasonografía, solicitadas en nuestro servicio, no le fue realizada la tomografía óptica coherente por tratarse de un paciente de un nivel socioeconómico bajo y la dificultad para trasladarse de Veracruz a la Ciudad de México. La tomografía axial computada y el electroencefalograma fueron solicitados por el servicio de neurocirugía, en el cual también se le daba seguimiento. El diagnóstico diferencial debe realizarse con melanoma amelanótico, hemangioma coroideo, linfoma coroideo primario, escleritis posterior, y metástasis coroidea.
El osteoma coroideo es intratable, su principal complicación, la neovascularización coroidea, puede responder a la fotocoagulación, terapia fotodinámica y a agentes antiangiogénicos.8
¿ Conclusiones
El osteoma coroideo es un tumor intraocular benigno poco frecuente, más común en pacientes jóvenes sanas. Nuestro paciente es un caso raro de osteoma coroideo en un varón de 9 años de edad, con antecedente de síndrome de Crouzon e histiocitosis de células de Langerhans remitida.
Correspondencia: Dra. Katia Calderón.
Insurgentes 3700, Col. Insurgentes Cuicuilco, Coyoacán. 04530, México, D.F.
Tel: 1084 0900.
Correo electrónico: katia_ csoto@hotmail.com, pediatria_inp@prodigy.net.mx
