


¿ INTRODUCCIÓN
El carcinoma suprarrenal, es una neoplasia maligna, agresiva, de mal pronóstico. Es un tumor raro que representa 0.02% de todas las neoplasias. La incidencia global es del 0.5 a 2 por 1000 000 de habitantes y la de los incidentalomas adrenales de 2% a 3%. Los no funcionantes predominan en la cuarta y séptima década de vida, con predominio masculino. La etiología es desconocida. El diagnóstico se realiza en la mayoría de las veces en estadios avanzados en forma incidental, metastatizan precozmente por su tendencia de invasión a estructuras vasculares.
La comprensión de la fisiología esencial de las glándulas suprarrenales se ha desarrollado a partir de la descripción inicial por Eustachius en 1563, con un análisis bioquímico suprarrenal secretor de productos y técnicas de imagen precisas, estudios actualmente disponibles. El reconocimiento de la presencia de las glándulas suprarrenales y su división en corteza y médula no fue hasta las observaciones de Addison, en 1855, cuando se observó la función esencial de estas glándulas en los pacientes que fallecieron con la destrucción suprarrenal secundaria a la tuberculosis. Poco después, realizó en animales adrenalectomías bilaterales y predijo que las glándulas suprarrenales son esenciales para la vida.1 La hiperfunción de la corteza suprarrenal no fue documentada hasta 1912, con un informe definitivo en 1932 de 11 pacientes con adenomas basófilos de la hipófisis que describen el ahora clásico síndrome de Cushing. Las formas de hipertensión adrenocortical, la hiperplasia suprarrenal congénita, carcinoma suprarrenal y otros trastornos suprarrenales.2-4
¿ PRESENTACIÓN DEL CASO CLÍNICO
Hombre de 43 años de edad, que acudió al servicio de urología referido por el de gastroenterología. Carga genética oncológica, madre con cáncer de mama, padre con cáncer de próstata, hermana con cáncer de ovario. Antecedentes personales no patológicos con tabaquismo positivo, etilismo social sin llegar a embriaguez, el resto sin relevancia. Portador de hipertensión arterial de 10 años de evolución, controlado con inhibidores de la angiotensina. Padecimiento actual de un año de evolución, con dolor abdominal postprandial, epigástrico e hipocondrio derecho, picos de dolor intenso con vómitos de característica gastrobiliar, es tratado por el servicio de medicina interna como enfermedad ácido péptica con pobre respuesta al tratamiento médico.
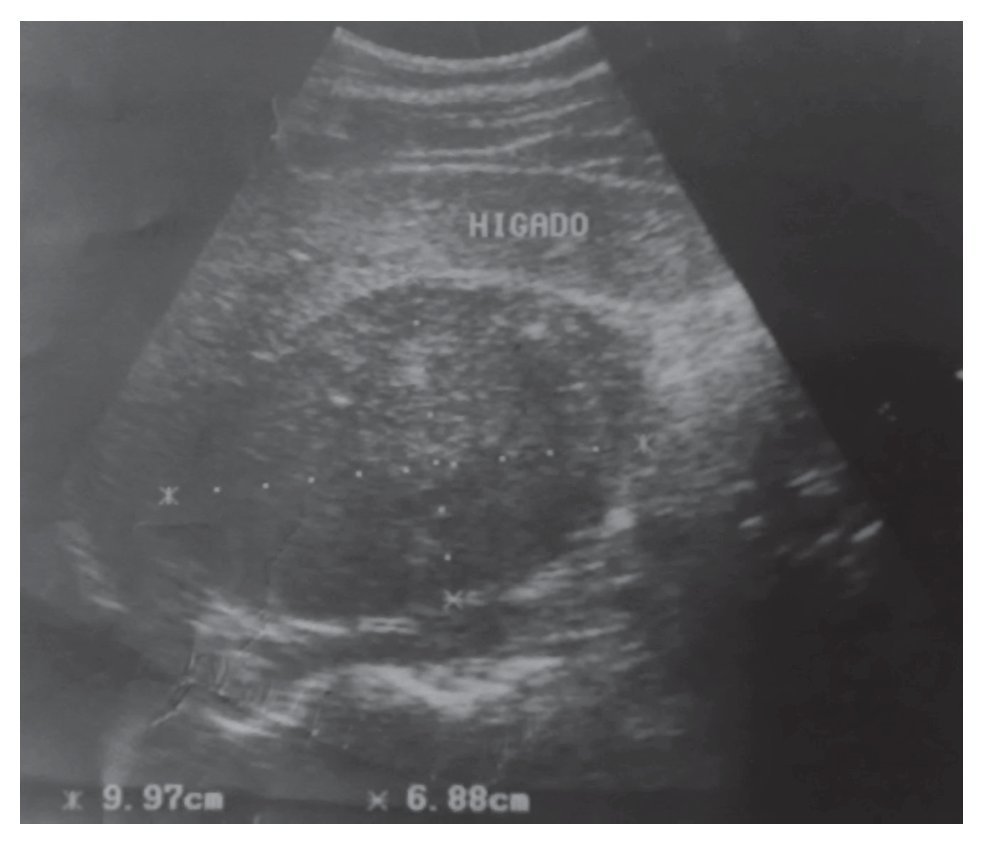
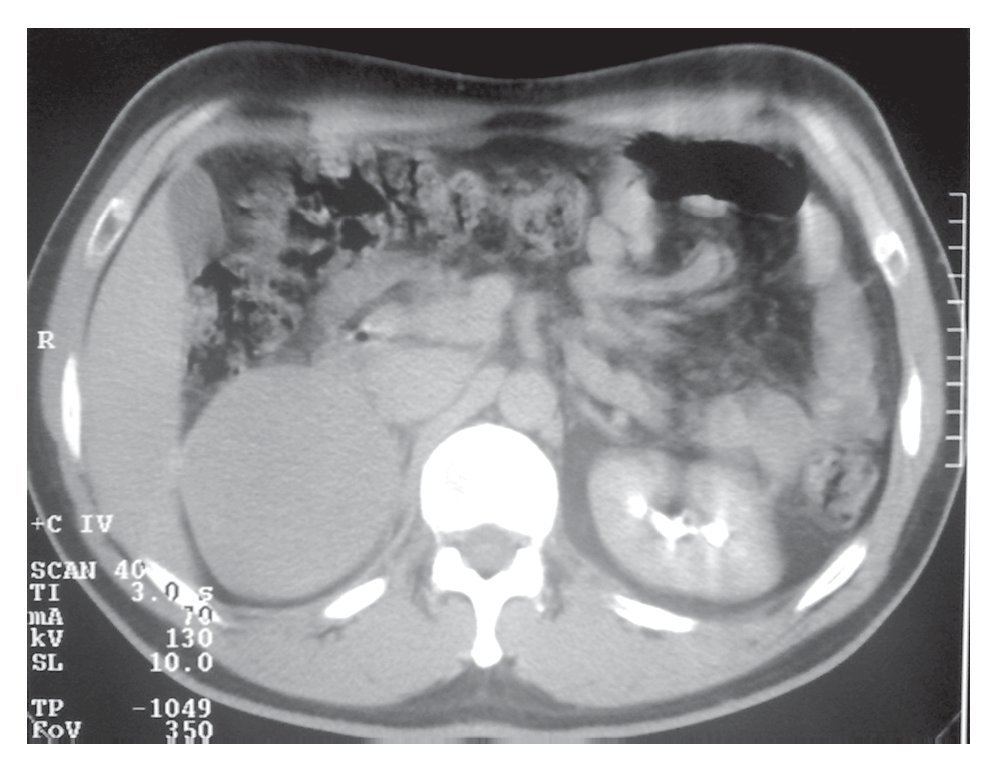
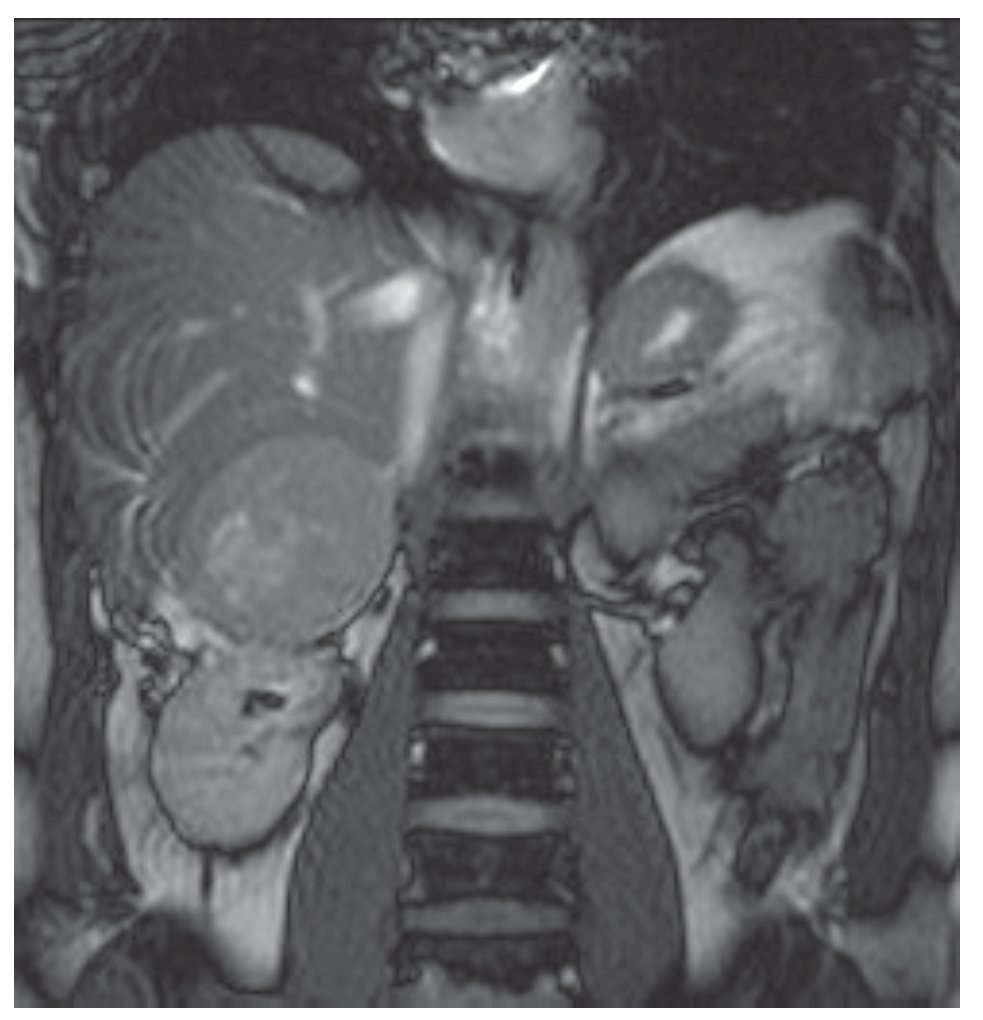
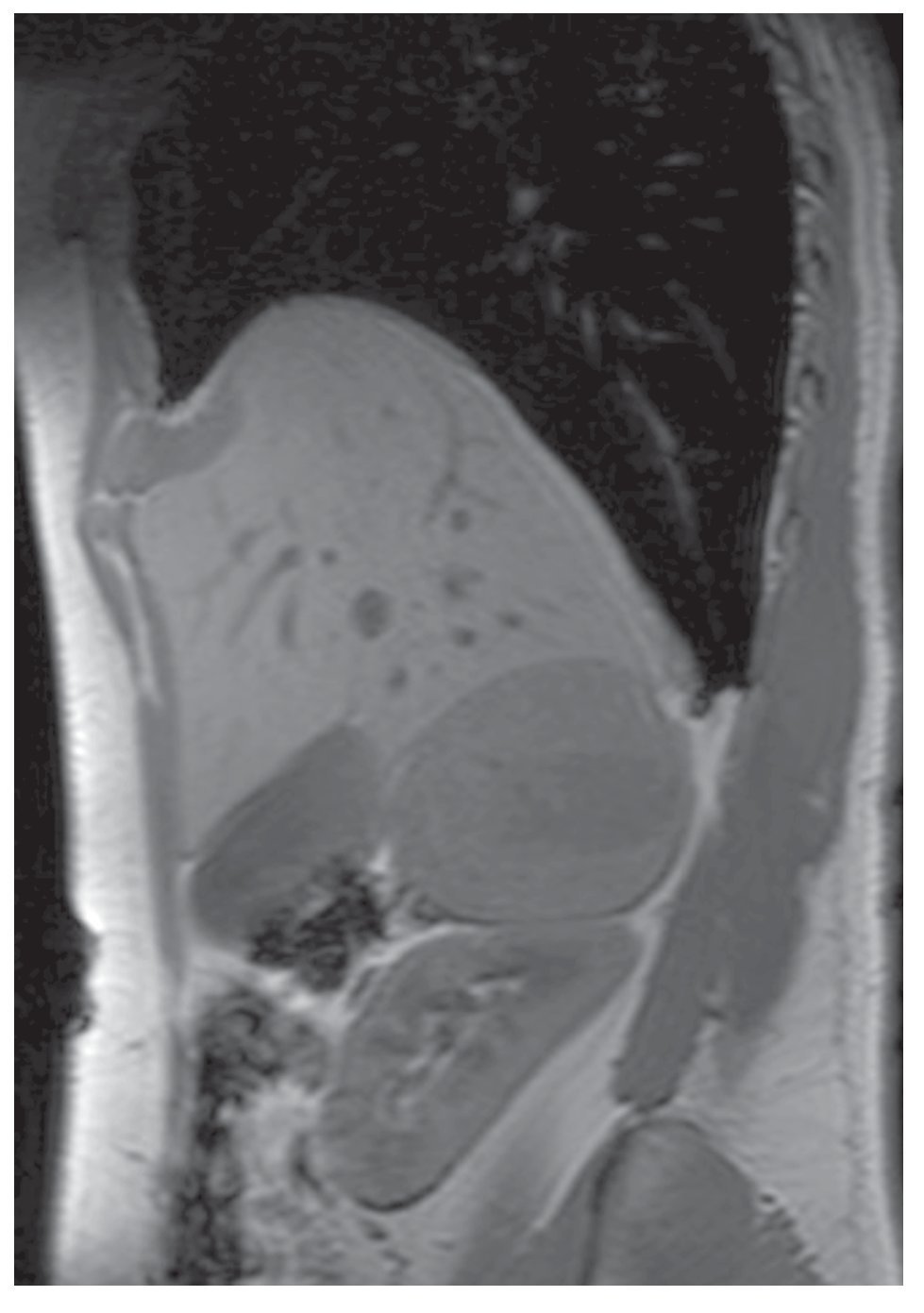
Su exploración física con signos vitales normales, peso de 107 Kg, talla de 184 cm. El resto sin datos relevantes. Se detectó masa suprarrenal derecha por ecografía. Laboratorio con biometría hemática, química sanguínea, electrolitos séricos, pruebas de función hepática, cortisol, aldosterona, normetanefrinas, metanefrinas en orina y totales normales. Gabinete: Ecografía con presencia de masa suprarrenal derecha heterogénea de 9.9 cm por 6.8 cm, confirmado con tomografía y resonancia magnética (Figuras 1, 2, 3 y 4).
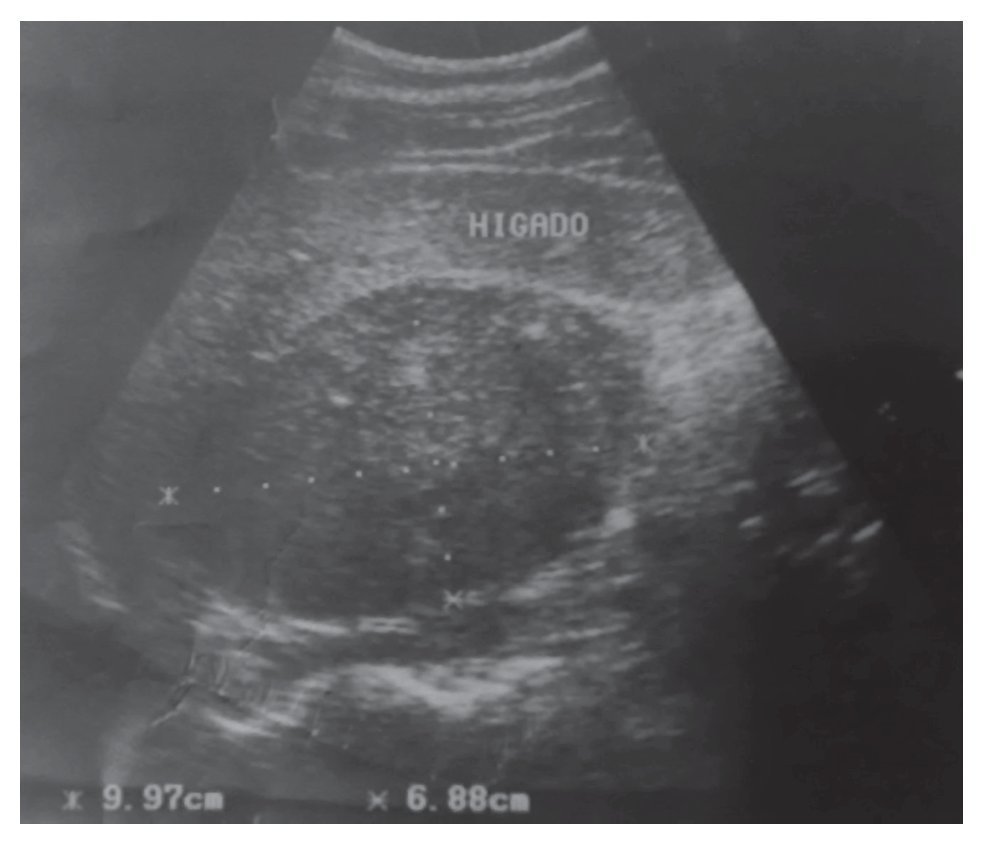
Imagen 1. Ecografía en la que se detecta la masa (inicialmente con sospecha de ser dependiente de hígado o vesícula biliar).
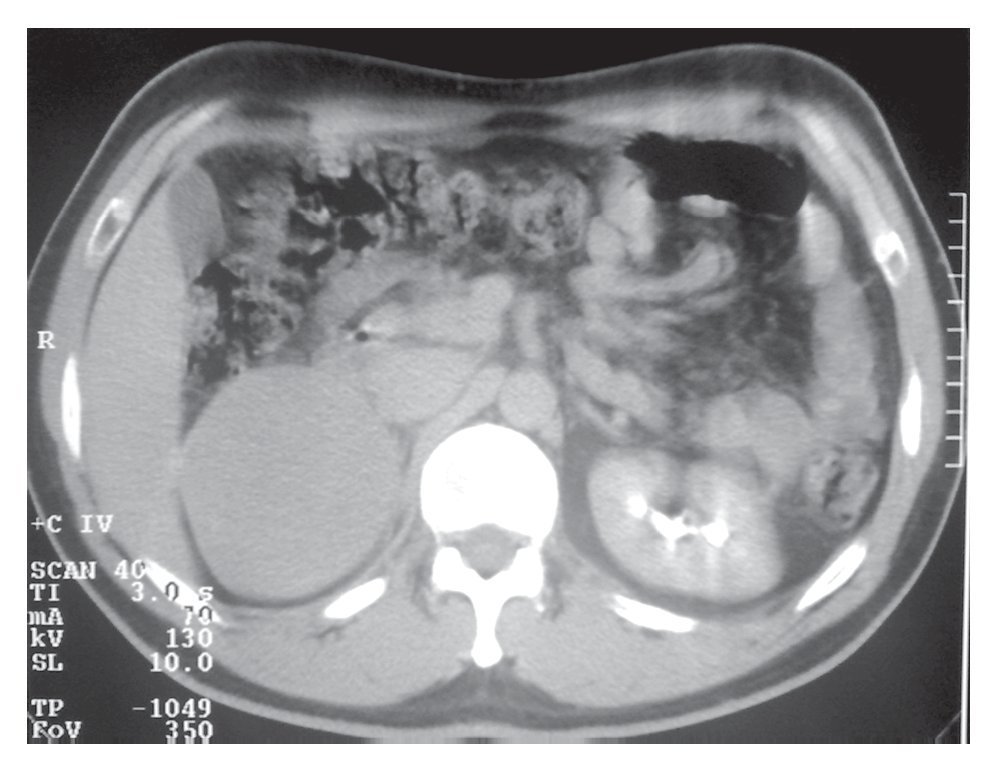
Imagen 2. Tomografía con medio de contraste endovenoso, no hay un reforzamiento de la masa en relacion a la TAC simple.
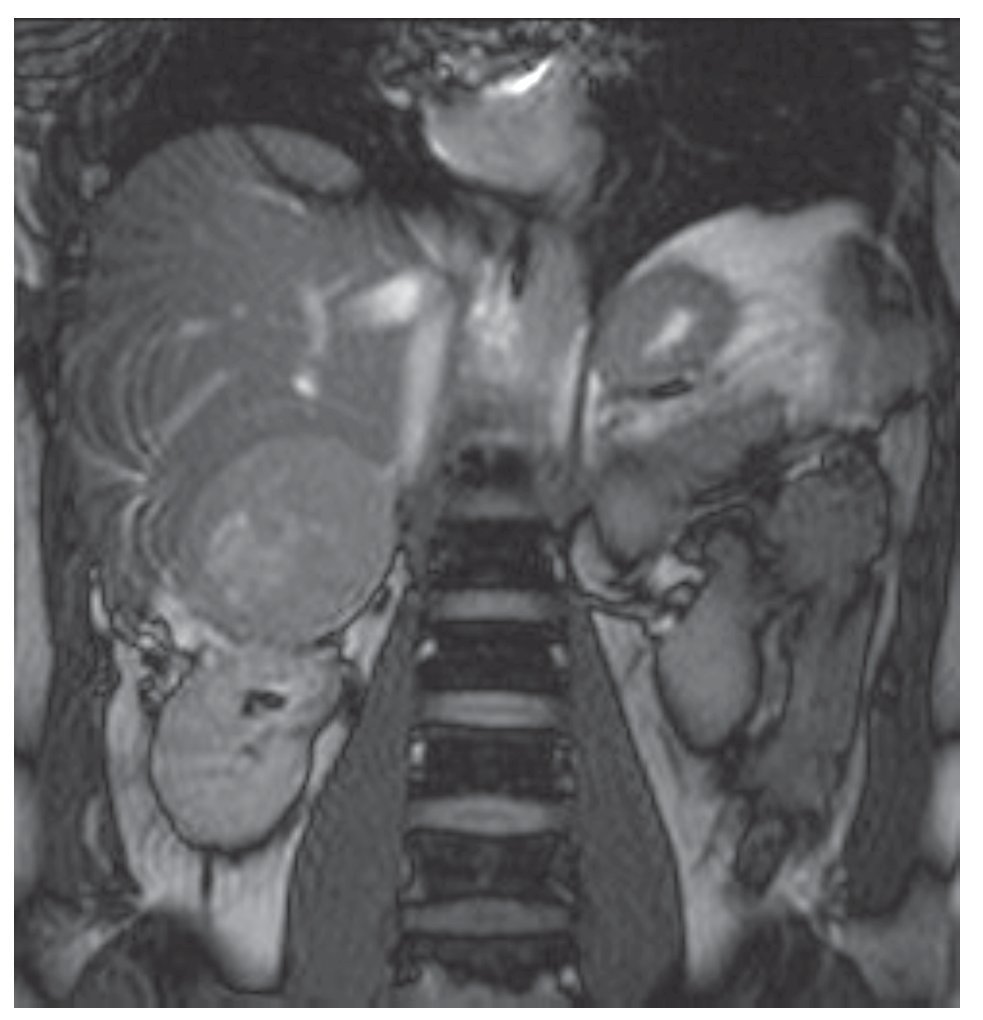
Imagen 3.Resonancia magnética, se evidencia masa dependiente de suprarrenal derecha, bien delimitada.
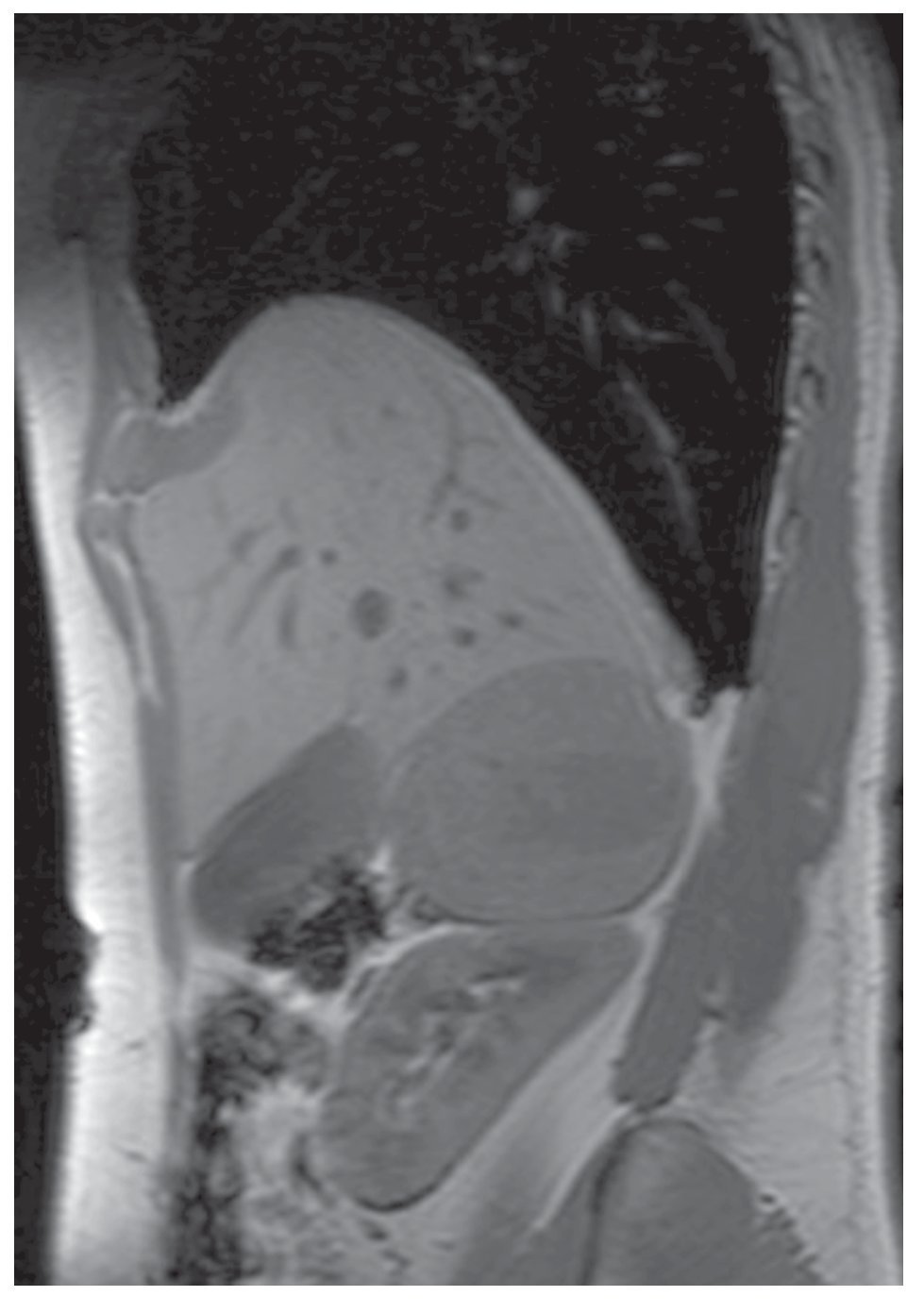
Imagen 4.Resonancia magnética.
Tratamiento: Ante los hallazgos clínicos y de gabinete, se diagnosticó masa suprarrenal derecha, se decidió realizar suprarrenalectomia derecha por laparoscopía con hallazgos quirúrgicos de tumor suprarrenal de 10 cm aproximadamente, bien encapsulado, sin evidencia macroscópica de invasión local. Se egresó a las 24 horas del posoperatorio, en buenas condiciones clínicas.
El resultado histopatológico al aspecto macroscópico señaló: espécimen de aspecto nodular de 261 g bien encapsulado de 9 cm por 7 cm por 5 cm, con áreas congestivas. Al microscopio se calificó con los criterios de Weiss con cinco puntos de nueve.
La evolución clínica del paciente a tres meses de egresado (y de realizar el presente trabajo) ha sido con buena y completamente asintomático.
Carcinoma suprarrenal: El carcinoma suprarrenal es un tumor poco frecuente, y los reportes en la literatura mundial son la mayoría estudios de casos. El diagnóstico se fundamenta en aspectos bioquímicos del síndrome hormonal y en las técnicas de imagen.5
Los incidentalomas suprarrenales tienen una prevalencia de 2.3% en la autopsia y de 0.5% a 2% en la tomografía abdominal.6 Un número reducido de tumores suprarrenales son funcionales y un número aún menor (1%) son de extirpe maligna. Las lesiones adrenales generalmente pasan clínicamente desapercibidas por cursar asintomáticas. La ecografía y la tomografía computarizada han incrementado notablemente los hallazgos y el diagnóstico, estas son las técnicas de diagnóstico con las que cuenta la mayoría de los hospitales, aun hay más herramientas de diagnóstico. La tasa de incidencia del carcinoma suprarrenal reportada es <0.03% de todos los incidentalomas suprarrenales que son de 1.5 a 6 cm de diámetro. El carcinoma suprarrenal, es un tumor maligno que representa 0.02% de todas las neoplasias malignas reportadas anualmente en los Estados Unidos de Norteamérica,7 con una incidencia de un caso por 1.7 millones de habitantes por año.8 El carcinoma suprarrenal en los niños apenas alcanza una incidencia de 0.002%. En algunas series la edad media del diagnóstico descrita en la literatura médica es de 50 a 60 años, pero según la edad de presentación para el sexo, en los varones es mayor respecto a las mujeres, acorde con la mayoría de las series publicadas.9 La mayoría de los tumores suprarrenales son esporádicos y unilaterales, pero 2% y 6% son bilaterales y se asocia con síndrome de Li-Fraumeni, la neoplasia endocrina múltiple tipo I, síndrome de Beckwith-Wiedemann, y el complejo de Carney, principalmente en los niños.10
Los tumores funcionantes son más frecuentes en mujeres y los síntomas de hiperfunción suprarrenal más frecuentes son el síndrome de Cushing y virilización por orden de frecuencia. Los tumores no funcionantes predominan en varones y generalmente se manifiestan como síndrome general o masa abdominal.11
El carcinoma suprarrenal tiene una edad de distribución bimodal, con un aumento en la incidencia en niños menores de cinco años de edad y en adultos en su cuarta y quinta décadas de la vida. El CAC no tiene predilección racial. Las mujeres elevan la proporción de aproximadamente 4:3. Los pacientes del sexo masculino tienden a ser mayores y tienen un peor pronóstico. Los adultos generalmente se presentan con etapas avanzadas de la enfermedad y la supervivencia de cinco años sigue siendo sombría, variando entre 16% y 38%.
La resección completa todavía ofrece la mejor oportunidad de cura, aunque la tasa de recurrencia es aún elevada y es estimada en 70% a 80%.12 La etiología es desconocida. La heterocigosidad cromosómica en los cromosomas 11p, 13q, 17p y pueden desempeñar un papel. Además, se han notificado anormalidades de p53.13
Anatomía suprarrenal: Las glándulas suprarrenales son órganos retroperitoneales, pares que se encuentran dentro de la grasa perinefrica en la cara anterosuperior y medial de los riñones. Miden hasta cinco centímetros de longitud por tres centímetros de ancho y son de un centímetro de espesor. Pesan aproximadamente cinco gramos cada una. En cambio, el peso al nacer suprarrenal es grande (de cinco a 10 g), a causa de la corteza suprarrenal fetal, que puede desempeñar un papel importante en la embriogénesis del feto y de la homeostasis. La corteza suprarrenal fetal involuciona rápidamente durante las primeras seis semanas de vida; por la muerte celular apoptótica en el momento del nacimiento del feto. Ambas glándulas son aplanadas en su cara anterior con una gruesa y delgada cresta central, medial y lateral.
La suprarrenal derecha se encuentra por encima del riñón, posterolateral a la vena cava inferior. La superficie anterior está en contacto inmediato con la superficie inferior y posterior del hígado. La vena cava inferior está medial; la superficie posterior de ambas glándulas suprarrenales está en contacto con la parte posterior del diafragma. La arteria renal izquierda se encuentra a menudo profunda a la vena suprarrenal. La glándula izquierda esta en el polo superior del riñón, con su superficie anterior y medial, detrás de la arteria esplénica y el páncreas. La superficie anterior de la suprarrenal izquierda puede estar expuesta por el resto de estructuras retroperitoneales y por el bazo. La glándula suprarrenal tiene un suministro de sangre de seis a siete mililitros por minuto. El drenaje venoso derecho es usualmente saliendo del vértice de la glándula y entra en la superficie posterior de la vena cava inferior; este sentido es breve, frágil y la fuente más común de sangrado molesto durante adrenalectomía derecha. La vena izquierda desemboca directamente en la vena renal izquierda, a unos tres centímetros de la vena cava inferior y a menudo frente a la vena gonadal.14-16
Embriología suprarrenal: La corteza suprarrenal se desarrolla a partir de mesodermo y la médula se desarrolla a partir del neuroectodermo. Durante la quinta semana de desarrollo, las células mesoteliales proliferan e invaden el mesénquima. Estas células forman la corteza del feto, un tipo de células es de origen mesonefrico. La relación íntima con el desarrollo de las gónadas, riñón y suprarrenal en general, explica el hallazgo ectópico o aberrante de tejido suprarrenal. El tejido ectópico se encontró en 2.7% de las exploraciones en la ingle los hijos varones y ninguno detectado en las mujeres. El examen microscópico de la corteza suprarrenal se divide en tres zonas: zona glomerular, zona fascicular y la zona reticular. La glomerular es menos prominente en los seres humanos que en otras especies y es el sitio de producción de aldosterona. La fascicular y la reticular son la forma funcional única que produce glucocorticoides, andrógenos y la médula suprarrenal estrógenos. La médula produce adrenalina y noradrenalina en presencia de los glucocorticoides. Además, existe evidencia morfológica de una estrecha interacción con las células cromafines de las células corticales, lo que sugiere un posible papel paracrino en la neurorregulación de la corteza suprarrenal.
Fisiología suprarrenal: La suprarrenal puede ser pensada funcionalmente como dos órganos, la corteza y médula. Cada una tiene su fisiología particular y sus propios productos secretorios de activación hormonal.
Corteza suprarrenal: La corteza suprarrenal produce una serie de hormonas esteroides que tienen una serie de acciones, incluyendo la retención de sal, la homeostasis metabólica y el desarrollo sexual.
La zona glomerular es la única fuente de los principales mineralocorticoides como aldosterona, que regula la reabsorción de sodio en el riñón, intestino, y glándulas sudoríparas y salivales. Las otras zonas pueden producir y secretar cortisol, el principal glucocorticoide en el ser humano, y los principales andrógenos dehidroepiandrosterona (DHEA), sulfato de DHEA (DHEAS) y androstenediona. El paso inicial para la formación de estas hormonas es la producción de pregnenolona.
El exceso de uno o varios productos de esteroides da signos y síntomas de síndrome de Cushing, hiperaldosteronismo primario (síndrome de Conn) o carcinoma suprarrenal. La regulación de la liberación de corticosteroides supone una compleja interacción del hipotálamo, glándula pituitaria y la glándula suprarrenal.
La hormona liberadora de corticotropina (CRH) se sintetiza en el hipotálamo y se lleva a la hipófisis anterior por la sangre. La CRH es un péptido lineal de 41 aminoácidos que estimula la liberación de ACTH. Por último, la secreción de ACTH, está mutuamente relacionada con el nivel de cortisol circulante. La producción de andrógenos suprarrenales en la zona reticular y fascicular también está bajo la influencia de la ACTH. En contraste con glucocorticoides y andrógenos suprarrenales, el principal control fisiológico en la secreción de aldosterona es la angiotensina II. El control de la ACTH es secundario.
La liberación de renina y la formación de angiotensina II, condiciona la secreción de aldosterona, lo que resulta en la retención de sodio en un intento de restaurar la perfusión renal. Por el contrario, si existe retención de sodio, se suprime la secreción de renina, la secreción de aldosterona se cae, y se incrementa el sodio urinario. Un segundo, pero menos potente estímulo para la liberación de aldosterona es el potasio.17,18
Acciones hormonales: Los glucocorticoides son esenciales para la vida, incluso después de la sustitución mineralocorticoide. Los glucocorticoides ejercen sus efectos sobre un amplio espectro de metabolismo celular, incluyendo la acumulación de glucógeno en el hígado y el músculo, el aumento de la gluconeogénesis, el deterioro de la utilización de glucosa periférica, atrofia muscular y la miopatía, la osteopenia inmune mediada por la inflamación y numerosas interacciones con otras hormonas.
La aldosterona representa 95% de la actividad suprarrenal mineralocorticoide y sirve para mantener el equilibrio del sodio y del potasio. Los sitios activos incluyen el riñón, intestino, glándulas salivales, y glándulas sudoríparas. En todos los sitios, el efecto es de estimular la reabsorción de sodio y el aumento de la secreción de potasio y el hidrógeno a través de la activación de la Na+-K+-ATPasa en la membrana19
Metabolismo: En la práctica, 80% de cortisol se une a la globulina corticosterona, de 10% a 15% se une a la albúmina y 7% a 10% es libre. Las elevadas concentraciones séricas de testosterona y DHEA son características de los tumores suprarrenales, que en las mujeres se presenta con hirsutismo.20
Médula suprarrenal: Está compuesta de grandes células cromafines, que secretan principalmente epinefrina, pero también norepinefrina y la dopamina. La enzima feniletanolamina-N-metiltransferasa (PNMT), que cataliza la metilación de la norepinefrina a epinefrina, está casi exclusivamente localizada en la médula suprarrenal. Si hay exceso de producción tanto de la norepinefrina y la epinefrina, la lesión es casi siempre dentro de la suprarrenal y no en otros sitios de tejidos cromafines. Estudios en seres humanos sanos indican que en plasma la dopamina representa 13% de la catecolamina libre, la epinefrina 14% y la norepinefrina 73%.
La estimulación de los nervios simpáticos preganglionares durante el estrés, dolor, frío, calor, asfixia, hipotensión, hipoglucemia y agotamiento de sodio, aumentan la liberación de catecolaminas.21
Metabolismo de las catecolaminas: Las catecolaminas son rápidamente eliminadas de la circulación, con una vida media en plasma de menos de 20 segundos. Las catecolaminas se degradan por la acción de la catecol-O-metiltransferasa y la monoamino-oxidasa, o bien con la enzima a partir de la degradación del proceso. El principal metabolito en la orina es acido vanillilmandelico; acompañado de metanefrinas, normetanefrinas y sus derivados, que a menudo se miden en la evaluación de los pacientes con feocromocitomas.22
Acciones de las catecolaminas: Las catecolaminas circulantes actúan en diferentes órganos con receptores específicos, condicionando la diversidad de los síntomas expuestos por los pacientes con feocromocitomas. Además, los distintos tumores pueden producir diferentes cantidades de norepinefrina, epinefrina o dopamina.
Cuadro clínico: La clínica de los carcinomas adrenales debe ser considerada en tres categorías: a) síntomas debidos a la propia masa adrenal, b) síntomas debidos a la diseminación locorregional, a distancia o ambos y c) síntomas endocrinos de hiperfunción con sobreproducción de hormonas activas o inactivas.
Aproximadamente 60% del carcinoma adrenal es hormonalmente funcional.23 Aunque varias hormonas esteroideas producidas son liberadas por las lesiones orgánicas, las más comunes son la hormona cortisol, aldosterona y los esteroides sexuales, lo que condiciona síndromes clínicos bien definidos. La mayoría de los carcinomas adrenales pueden secretar varias hormonas y pueden cambiar la secreción según el tamaño, la tasa de crecimiento y la diferenciación histológica.
El carcinoma adrenal tiene una presentación clínica común: el exceso de glucocorticoides, visto en alrededor de 45% de los casos, o una mezcla de Cushing y el síndrome virilizante con exceso de glucocorticoides y andrógenos, lo que representa 25% de los casos.24-26
El Síndrome de Cushing es un complejo grupo de síntomas funcionales derivados de los tumores con exceso de cortisol. Los síntomas varían, pero la mayoría de los pacientes tienen obesidad troncular, cara redondeada y adelgazamiento en los brazos y las piernas. La piel se convierte en delgada y frágil con frecuentes equimosis. Pueden aparecer estrías cutáneas en el abdomen, muslos, glúteos, brazos y pechos. Muchos pacientes tienen fatiga, debilidad muscular proximal, osteoporosis, hipertensión, glucosa e intolerancia; manifestaciones de irritabilidad, ansiedad y depresión pueden ser frecuentes. Las mujeres normalmente tienen hirsutismo, dismenorrea o amenorrea y rara vez clitoromegalia. En los hombres es frecuente disminución de la fertilidad o ausencia de la libido. Tiende a ser más marcada la virilización en Cushing y virilización en mujeres y feminización de los hombres que tienen el CAC tales como: ginecomastia, atrofia testicular y bajo recuento de esperma; se ha observado en pacientes con tumores secretores de androstenediona periféricamente convertidos a estrógenos.24
Hay una amplia lista de diagnóstico diferencial que incluye: Síndrome de Cushing o el síndrome virilizante. La mayoría de los pacientes con carcinomas no funcionantes se presentan con signos y síntomas relacionados para el crecimiento del tumor, tales como dolor abdominal o en flanco o plenitud. Comúnmente los pacientes se pueden presentar con una masa suprarrenal, que se encontró incidentalmente en técnicas de imagen realizadas por una razón diferente.
Clasificación clínica: Una práctica subclasificación de los carcinomas adrenales está de acuerdo con su capacidad de producir hormonas adrenales. La mayoría de los tumores segregan múltiples compuestos. Una revisión de 62 pacientes con tumores funcionales informó que 53% tenía síndrome de Cushing, virilización sólo 21%, 10% síndrome de Cushing y virilización, feminización 8% y 5% hiperaldosteronismo.26
Hallazgo incidental de masas adrenales: El carcinoma adrenal cortical representa 2% de los tumores que son menores de cuatro centímetros, 6% de los tumores que son 4.1 a 6.0 cm y 25% de los tumores que son más grandes que 6.0 cm. Los tumores malignos suelen ser mayores de seis centímetros. En consecuencia, las lesiones adrenales sólidas de más de 6 cm deben ser consideradas malignas hasta que se demuestre lo contrario por exploración y adrenalectomía.
El seguimiento de los pacientes con nódulos menores revela que 5% a 25%, aumentó en su tamaño en al menos 1 cm durante un tiempo variado, el riesgo de malignidad es de uno de 1000 y hasta 20% pueden desarrollar hiperfunción hormonal, especialmente si el tumor es mayor de tres centímetros.27,28
Todos los pacientes con masas adrenales sólidas deben someterse a evaluación bioquímica. Se recomienda la evaluación, incluyendo orina y plasma con determinación de metanefrinas para descartar feocromocitoma; también la supresión de esteroides para descartar síndrome de Cushing y, en pacientes con hipertensión arterial, medir el potasio sérico, la concentración de renina plasmática y la proporción de aldosterona.
Por la probabilidad de que las lesiones no funcionantes sólidas de más de seis centímetros sean malignas, estas lesiones deben ser eliminadas. La TAC puede subestimar el tamaño de una lesión suprarrenal, por lo que se sugiere la exploración quirúrgica cuando la lesión es mayor de cinco centímetros en la TC o RM.
Hay discrepancia entre varios autores, en los que toman rangos de 4.0 cm (±1) para decidir la intervención quirúrgica o la vigilancia conservadora. Otros autores incluyen la eliminación de tumores más pequeños (3 y 4 cm) en pacientes menores de 50 años, también se propone un abordaje quirúrgico, especialmente en los pacientes más jóvenes.28
Diagnóstico, evaluación y manejo preoperatorio: La evaluación endocrina cuidadosa antes de la cirugía es crucial para el carcinoma suprarrenal. El patrón de secreción hormonal puede señalar al potencial maligno de la lesión y puede afectar la estrategia quirúrgica. La secreción autónoma de cortisol por el tumor se asocia con el riesgo de insuficiencia suprarrenal en el postoperatorio. Un manejo endocrino es importante en el establecimiento de marcadores tumorales para el seguimiento recidiva tumoral. Tanto el tamaño como la apariencia de la masa suprarrenal de TC, RM y más recientemente, la 18F-fluorodesoxiglucosa, así como tomografía por emisión de positrones (PET-FDG), se han utilizado para distinguir entre lesiones benignas y malignas. Ninguna de estas pruebas de imagen son específicos para el carcinoma suprarrenal, pero cada modalidad es complementaria a las demás.
El tamaño de la masa suprarrenal, medida por el TC o RM, sigue siendo uno de los mejores indicadores de potencial maligno. Masas suprarrenales mayores de seis centímetros son muy sospechosas de malignidad y deben ser removidas quirúrgicamente. Masas adrenales inferiores a tres centímetros son menos propensas a ser malignas y pueden ser objeto de seguimiento. Los tumores entre tres y seis centímetros de diagnóstico representan el principal desafío.
La sensibilidad de la RM para diferenciar tumores benignos y malignos varía entre el 81% y 89%, con una especificidad entre 92% y 99%. Algo útil característico de la RM es que puede identificar la invasión tumoral en la vena cava inferior (VCI) lo que permita mejorar la planeación quirúrgica.29,30
La aspiración con aguja fina se realiza sólo si el abordaje quirúrgico no es posible y el diagnóstico no puede ser establecido con imágenes TC, RM.
Un nuevo método de imagen suprarrenal es con metomidato-11C-PET. El metomidato suprarrenal se une a 11-b-hidroxilasa, por lo que es potencialmente una herramienta útil para distinguir las lesiones de origen adrenocortical de otras lesiones. Este método está disponible en la mayoría de los centros de investigación y más aún en centros de Europa.31-34 Es importante corregir las anormalidades de electrolitos que puede ser el resultado de un tumor suprarrenal endocrinológicamente activo. En particular, en el aldosteronoma la hipopotasemia puede dar lugar a requerir repleción de potasio y la administración de diuréticos ahorradores de potasio. La hipertensión arterial también debería ser tratada antes de la cirugía. En el feocromocitoma, el bloqueo α-adrenérgico debería comenzar dos semanas antes de la cirugía. La Fenoxibenzamina (10 mg a 40 mg cuatro veces al día, máximo, 300 mg/día) se utiliza, comenzando poco a poco con una dosis de 10 mg a 20 mg dos veces al día.
Algunos pacientes con taquicardia pueden beneficiarse de cuadros concurrentes con bloqueo b. Alternativamente, un bloqueador selectivo a1, como prazosina o doxazosina, puede ser utilizado. Intraoperatoriamente, la presión arterial elevada puede ser tratada con nitroprusiato o un β-bloquerador de acción corta como el esmolol. La repleción de volumen es importante para prevenir la hipotensión secundaria a la pospérdida de la vasoconstricción después de la eliminación de un feocromocitoma. En los pacientes con síndrome de Cushing se debe exigir la corrección de las anomalías de electrólitos antes de la cirugía, así como de la hiperglucemia. Estos pacientes pueden beneficiarse con la administración de agentes adrenolíticos como mito-tañe o aminoglutetimida.35
Una preparación mecánica del intestino es útil para cirugía abierta o cirugía laparoscópica transperitoneal. En la cirugía retroperitoneal no es necesaria la preparación intestinal. Todos los pacientes deben recibir antibióticos preoperatorios apropiados.
Tratamiento quirúrgico: Completar la resección quirúrgica es el único tratamiento potencialmente curativo de los pacientes con carcinoma adrenocortical. Otros estudios publicados han informado que la supervivencia a cinco años oscila entre el 32% a 48%, con una mediana de supervivencia de dos años para los pacientes sometidos a resección completa, en comparación con una mediana supervivencia de menos de un año para los pacientes sometidos a resección incompleta.36,37 La presencia de invasión en VCI no debería ser considerado como enfermedad metastásica, sino como extensión del tumor. En tales casos, el procedimiento quirúrgico debe ser más agresivo, tratando de eliminar por completo la lesión intravascular. La indicación de la escisión total del tumor en pacientes con estadio III y IV de la enfermedad sigue siendo polémica. El tratamiento quirúrgico paliativo no influye en la supervivencia de estos pacientes. La invasión del tumor en el eje del tronco celiaco, la aorta, o proximal a la arteria mesentérica superior representa clara evidencia de un tumor localmente no resecables.38
Adrenalectomía abierta: La adrenalectomía abierta se puede realizar por vía transperitoneal o retroperitoneal. Por línea media transperitoneal, subcostal, o toracoabdominal. Las ventajas de la transperitoneal son: mejor exposición de los tumores más grandes y excelente acceso. Las principales desventajas son: íleo prolongado y difícil exposición en el paciente obeso mórbido. El enfoque retroperitoneal da los mismos resultados excepto en íleo y puede dar lugar a estancias hospitalarias más cortas. Se ha recomendado la cirugía abierta para cualquier sospecha o certeza de malignidad adrenocortical, facilitar la máxima exposición de la resección quirúrgica completa y minimizar el riesgo de diseminación tumoral.
Adrenalectomía laparoscópica: Debido a la alta recidiva local y la difusión intraperitoneal, la adrenalectomía laparoscópica debería reservarse para la resección de presuntos tumores benignos corticales (<3 cm).39,40
Sin embargo, si la tomografía computarizada indica que no está presente la invasión local y la lesión no es excesivamente grande, la adrenalectomía laparoscópica es posible. El compromiso de la vena suprarrenal o la vena cava es una contraindicación absoluta para la cirugía laparoscópica. Actualmente la cirugía adrenal laparoscópica es considerada con excelentes resultados constituyéndose en el estándar de oro.
Adrenalectomía robótica: La cirugía robótica está ampliándose a realizar este tipo de intervenciones con resultados buenos, pero el costo es elevado.41
Ablación por radiofrecuencia: La ablación por radio-frecuencia se ha utilizado con éxito para tratar lesiones adrenales, sobre todo las metástasis suprarrenal o metástasis de carcinoma de la corteza suprarrenal.42
Crioablación: La experiencia clínica con éste recurso es bastante limitada. Experimentos con perros han confirmado que la crioterapia puede ser realizada en la glándula suprarrenal eficazmente.43
Radioterapia: Ha sido considerada por la mayoría de los expertos en el campo como ineficaz para el tratamiento del carcinoma suprarrenal. El uso de radioterapia se limita al tratamiento paliativo de tumor irresecable o a la enfermedad metastásica con pobres resultados.44,45
Quimioterapia: El mitotane es el único medicamento aprobado por la FDA para el tratamiento del carcinoma suprarrenal. Fue desarrollado en 1960 como un insecticida, el DDT, afecta la corteza suprarrenal, causando necrosis. Ejerce un efecto citotóxico sobre células adrenocorticales, produciendo degeneración focal de la zona fascicular y en particular de la reticular, mientras que los cambios en la zona glomerulosa son relativamente leves.
La actividad antitumoral se produce a una concentración plasmática de 14 mg/L o mayor, pero los efectos secundarios tóxicos resultan en el nivel superior a 22 mg/L.
Con frecuencia hay efectos adversos (reversibles) en el sistema gastrointestinal o el sistema nervioso central.
La administración concomitante de glucocorticoides es recomendada. El mitotane se ha utilizado en la fijación de los adyuvantes después de la terapia curativa o la resección quirúrgica completa. El promedio de tasa de respuesta objetiva del tumor al mitotane es de aproximadamente 32%.44
Quimioterapia citotóxica: La experiencia con la quimioterapia citotóxica en el carcinoma suprarrenal es limitada. No ha sido evaluada adecuadamente por la rareza de la enfermedad. La quimioterapia basada en cisplatino tiene actividad en el CAC. Varios regímenes de quimioterapia se han combinado con mitotane, Se han utilizado esquemas con mitotane-doxorrubicinaetopósido-cisplatino con una tasa de respuesta de 49% pero con toxicidad significativa. La tasa de respuesta con quimioterapia basada en cisplatino es de aproximadamente 20% a 30%. La combinación de mitotanestreptozotocina tiene una tasa de respuesta objetiva de 36%.46
Para realizar el plan terapéutico, en 1977 el Departamento de Salud de los Estados Unidos de Norteamérica (SEER) creó una clasificación, modificada por Sullivan en 1978, considerando cuatro estadios:
1. Enfermedad localizada. Tumor que tras su extirpación esta confinado a la adrenal, no pudiéndose poner de manifiesto la existencia de metástasis. Menos de 5% del carcinoma adrenal se encuentra en esta categoría.
2. Enfermedad Regional. Tumor con afectación de tejidos contiguos y de órganos vecinos, incluyendo ganglios linfáticos, sin metástasis. Entre 20% a 30% de los tumores pertenecen este grupo.
3. Enfermedad a distancia. Tumores metastatizados al momento del diagnóstico, en este grupo se encuentran 40% a 60% de los tumores
4. Enfermedad recurrente. Aparición de recidiva local o diseminación a distancia tras la reseccion teóricamente curativa del tumor primario.47
Metástasis: A excepción de los tumores secretores de testosterona, los carcinomas corticosuprarrenales son muy malignos, presentan diseminación tanto local como hemática, con tasas de supervivencia bajas. Los sitios más comunes de metástasis son los pulmones, el hígado y los ganglios linfáticos. Estudios de autopsias revelaron 132 casos de metástasis a los pulmones (60%), hígado (50%), ganglios linfáticos (48%), hueso (24%), pleura y corazón (10%). Además, estos tumores se extienden con frecuencia directamente en las estructuras adyacentes, especialmente en el riñón y pueden involucrar la vena cava o la vena esplénica.48
Pronóstico: La mayoría de los pacientes se presentan en etapas avanzadas de la enfermedad. En casi 70% de los casos de carcinoma adrenal la enfermedad se ha propagado más allá de la glándula suprarrenal en el momento del diagnóstico. Al diagnosticar, los pacientes que tienen la fase I y II de la enfermedad representan aproximadamente 32% de los casos y 68% actual en etapa III y IV de la enfermedad.
La resección completa ofrece una oportunidad de curación entre los pacientes que presentan tumores localmente avanzados y con enfermedad resecable. De los pacientes, 80% tienen recidiva local o metástasis después de la resección radical. Otras series de casos también informan de que la supervivencia global fue superior en los pacientes que respondieron a mitotane.
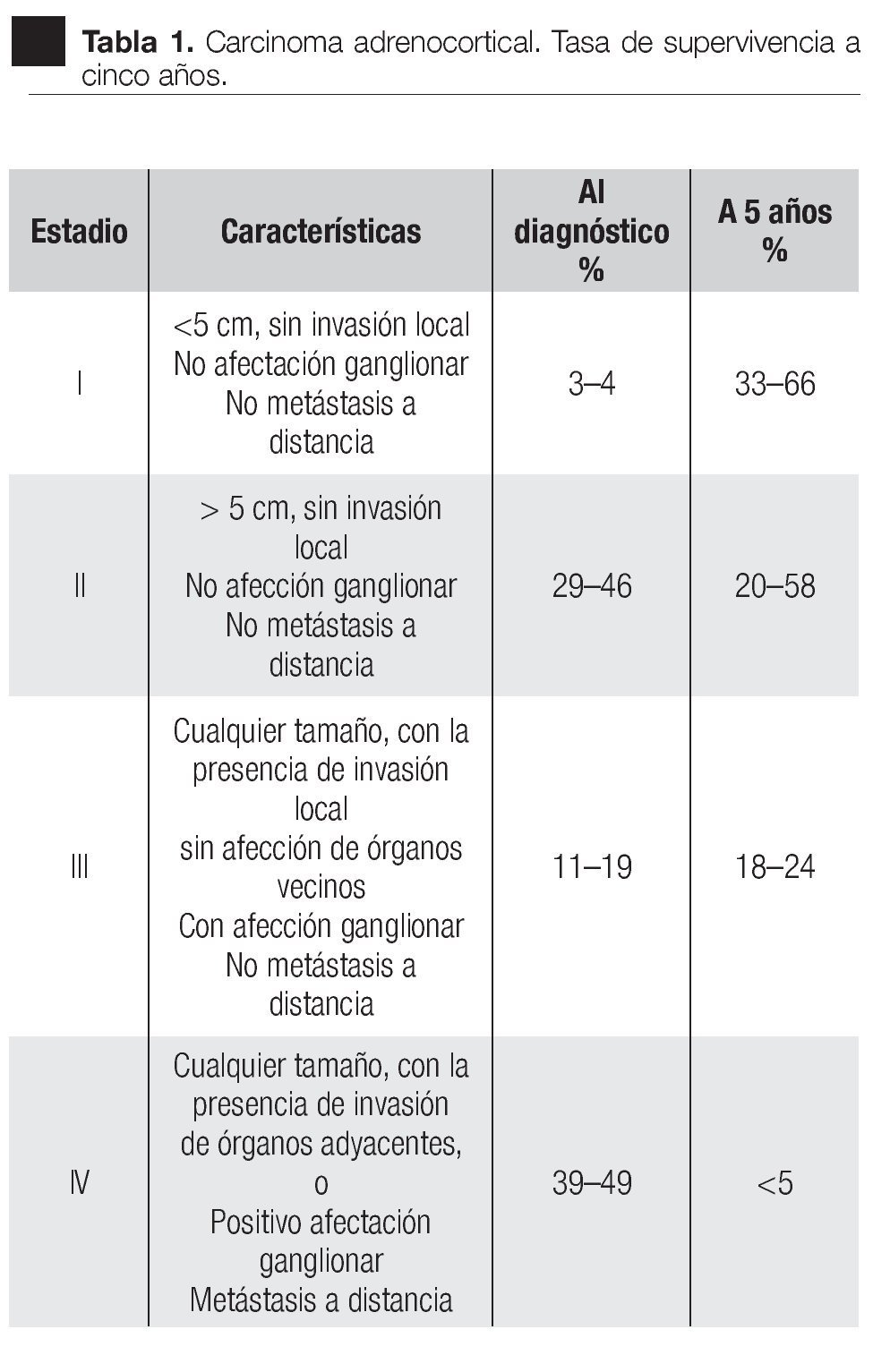
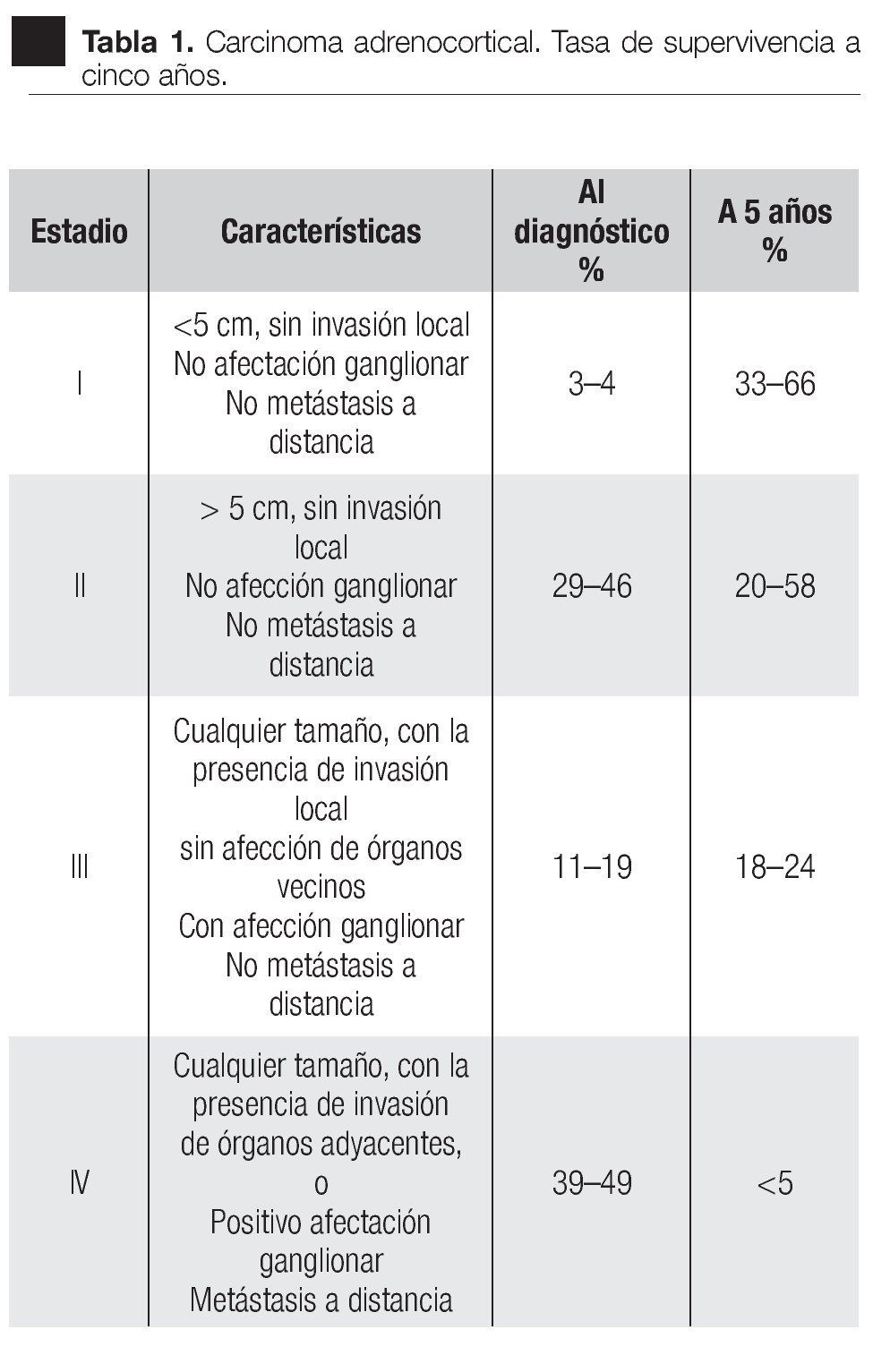
La rareza de la enfermedad ha limitado la acumulación de estudios adecuados para correlacionar el tamaño de la población o para validar cualquier factor pronóstico (molecular o clínico) entre los pacientes. Además, la presencia de metástasis a distancia, la edad avanzada (>45 años) y la resección incompleta fueron los factores determinantes de la supervivencia (Tabla 1).49
Diagnóstico histopatológico: El diagnóstico patológico debe ser realizado por un experto. La diferenciación entre tumores adrenales benignos y malignos es difícil y se basa en las características macroscópicas y microscópicas.
Macroscópicas: Valorar el peso, hemorragia, invasión tumoral cápsula, y la invasión vascular. La puntuación Weiss es la más ampliamente utilizada para la clasificación de las características microscópicas sugestivas de un tumor maligno y enumera nueve criterios histológicos:
1. Alta tasa de mitosis (>5 por 50 de alto campo).
2. Mitosis atípicas.
3. Invasión venosa.
4. Alto grado nuclear (Fuhrman 3 a 4).
5. Ausencia de células con citoplasma claro (<25% de las células).
6. Un patrón de crecimiento difuso (más de un tercio de tumor).
7. Necrosis.
8. Invasión sinusoidal.
9. Invasión capsular.
Son necesarios tres o más para realizar diagnóstico de carcinoma suprarrenal.50 Es importante la información adicional obtenida con inmunohistoquímica. Varios estudios han demostrado el valor de la tinción Ki67 en la diferenciación de lesiones benignas y malignas.51 El Melan-A (MART-1) en un gen que codifica un antígeno reconocido por células T citotóxicas. Aunque MART-1, se ha dicho ser restringido en su expresión a los melanocitos, los investigadores del Memorial Sloan-Kettering Cáncer Center han informado de que tiene un potencial de diagnóstico aplicable en el CAC. Una vez que el melanoma se excluye, la presencia de inmunorreactividad para el A103 (un anticuerpo a MART-1) excluye cualquier otra posibilidad de carcinoma que pueda entrar en el diagnóstico diferencial del carcinoma suprarrenal.
Otros marcadores, como la D11, inhibina-alfa, Melan-A y cromogranina-A, ayudan a definir si el tumor es adrenocortical o descartar un tumor de origen. Nuevos marcadores (LOH en 17p13, con sobreexpresión de IGF-2, ciclina E) se han propuesto para separar lesiones suprarrenales malignas de las benignas.52
Epidemiología internacional: En relación al carcinoma de glándula suprarrenal, no hay una información objetiva en los archivos de la Organización Mundial de la Salud. Es evidente que no se encuentra esta patología dentro de las primeras causas de enfermedad. Lo informado en la literatura médica mundial es una incidencia de un caso en 1.7 millones. Generalmente se reporta en adultos, con una media de 44 años, en etapa reproductiva, siendo éste de un potencial maligno agresivo con una tasa de supervivencia a cinco años alrededor de 40%.
Epidemiología nacional: No hay informes en la Dirección General de Epidemiología de la Secretaria de Salud de México. En México, sólo se cuenta con informes de casos esporádicos, como casos clínicos; se trabaja con lo señalado en la bibliografía mundial.
Correspondencia: Dr. Marco Antonio Vilchis Cárdenas.
Avenida Fidel Velázquez y Abraham Lincoln. Colonia Morelos. Monterrey, Nuevo León, México. Teléfono - Fax: (5281) 83714100, extensión 41315. Teléfono: (045) 8110 50 8553.
Correo electrónico: marcoavil@hotmail.com