


¿ INTRODUCCIÓN
El feocromocitoma es una neoplasia que se desarrolla de las células cromafines las cuales son derivadas del ectodermo neural y la mayoría se encuentran localizadas en el tejido adrenal.1
El paraganglioma representa menos de 0.05% del cáncer vesical que no depende del urotelio y 10% de la representación extra-adrenal del feocromocitoma; aproximadamente 15% son malignos, tiene una sutil predominancia hacia el sexo femenino.1
Desde 1953, menos de 200 pacientes han sido reportados en la literatura como casos de paragangliomas. El feocromocitoma maligno (definido como el feocromocitoma metastásico) se encuentra en 15% de los paragangliomas, con menos de 20 casos señalados en este contexto.2
Las mutaciones de las líneas germinales en los genes que codifican las subunidades B, C y D del complejo mitocondrial II succinato deshidrogenada (SDH) se asocian con múltiples paragangliomas. Estos genes SDH tal vez se comporten como genes supresores de tumores. Las mutaciones en el gen de la succinato deshidrogenasa D (SDHD)3 son la causa más frecuente de paragangliomas en cabeza y cuello (también llamados gliomas), mientras que las mutaciones en la succinato deshidrogenada B (SDHB) se relacionan con mayor frecuencia a feocromocitoma adrenal y extra-adrenal de características malignas.
En contraste, el paraganglioma maligno sólo se ha asociado ocasionalmente con mutaciones en el gen SDHD.4 El paraganglioma deriva de células croma-fines del plexo simpático del músculo detrusor; la liberación de catecolaminas puede ser impulsada por el vaciamiento, distensión vesical, defecación o el coito. Los síntomas típicos incluyen diaforesis, hipertensión paroxística, taquicardia, cefalea, y síncope.
Frente a la sospecha de paraganglioma vesical, la cistoscopia debe realizarse previo bloqueo adrenérgico. No es recomendable biopsiar estos tumores debido a que se encuentran hipervascularizados y generalmente cubiertos de urotelio normal.5
El tratamiento del tumor localizado es la resección transuretral.6 La cistectomía parcial se recomienda ante tumores de gran tamaño o ante una extensión ganglionar linfática. En 1988, Averbuch reportó la eficacia de la quimioterapia, demostrando respuestas favorables con marcadores bioquímicos negativos en 78% y margen de tumor negativo en 57%.7 De cualquier forma, no se ha estandarizado el tratamiento para feocromocitoma refractario a quimioterapia y la supervivencia media es de 16 meses desde el diagnóstico de las metástasis. Es conocido que el feocromocitoma maligno no responde a la radioterapia y ésta se limita a tratamiento paliativo de metástasis óseas, algunos estudios recientes proponen a la radioterapia como inhibitoria del crecimiento del tumor previamente irradiado (a pesar de la progresión del tumor fuera del tejido irradiado), disminuyendo el nivel de catecolaminas y probablemente sea efectiva para el tratamiento de enfermedad refractaria a quimioterapia.8
¿ PRESENTACIÓN DEL CASO
Mujer de 66 años con hija con glioma en el sistema nervioso central. Diabética e hipertensa en control con diferentes antihipertensivos. Inició su padecimiento en julio de 2008, cuando fue referida por médico particular a la consulta externa de urología del Hospital Central Militar por presentar tres eventos de hematuria macroscópica, silente, con coágulos amorfos, acompañado de cefalea, tinnitus, palpitaciones y diaforesis.
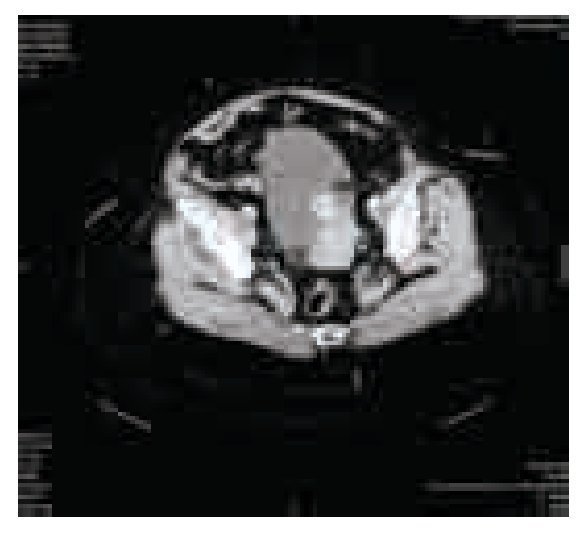
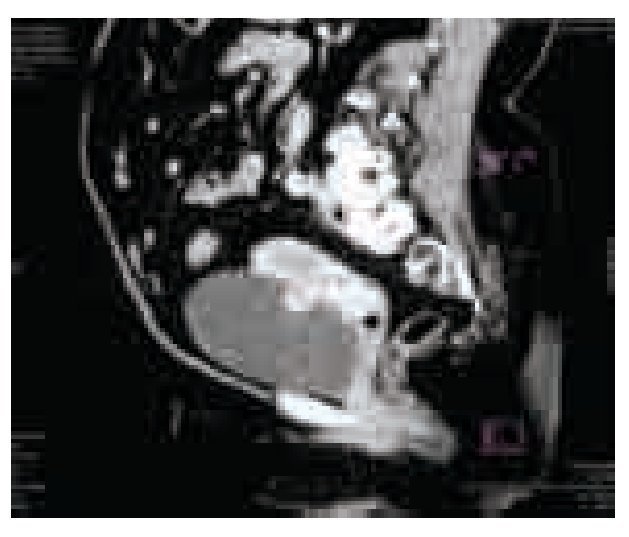
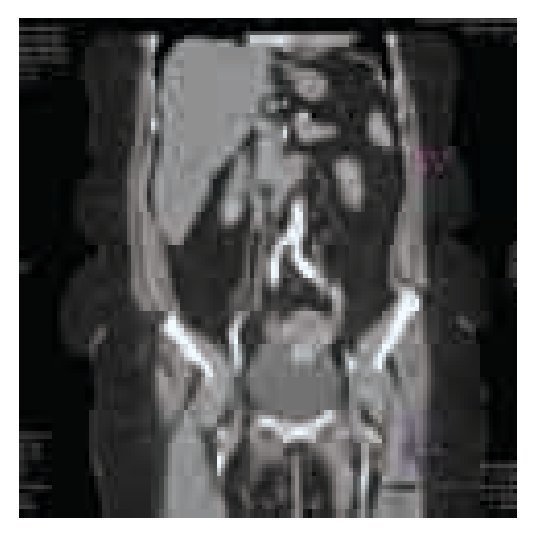
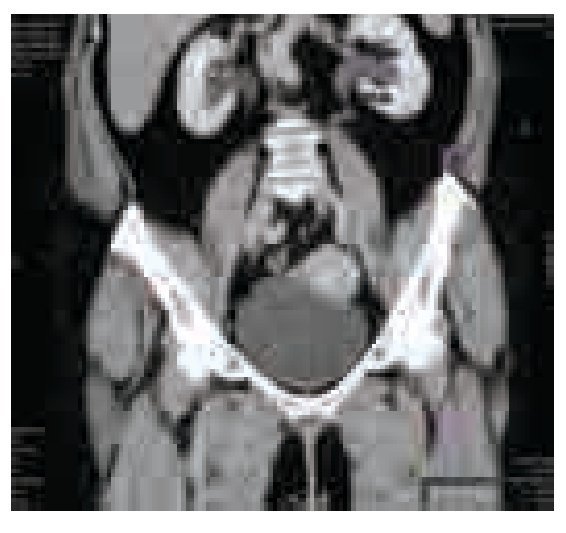
El examen general de orina resultó con hematuria, sin datos de infección. La cistoscopia identificó una tumoración pediculada, de bordes lisos, aspecto rosáceo de base amplia con abundante vascularización de aproximadamente 5 cm retrotrigonal, en izquierdo, (sin características macroscópicas del tipo de células transicionales) no se identificó sangrado activo, en estudio urotomográfico se observó masa vesical (Imágenes 1, 2 y 3) y ectasia del sistema colector de predominio izquierdo, sin elevación de azoados (Imagen 4).
Imagen 1.Corte axial de urotomografía abdominopélvica en su fase arterial en donde se observa imagen hiperdensa en piso vesical.
Imagen 2.Corte coronal de urotomografía abdominopélvica en su fase venosa; se repite imagen hiperdensa en piso vesical.
Imagen 3.Corte sagital de urotomografía abdomino-pélvica en su fase venosa, corroborándose imagen hiperdensa y su adecuada interfaz con el útero.
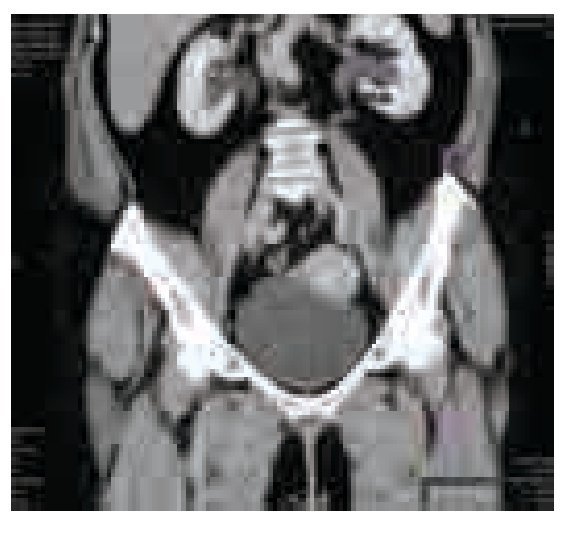
Imagen 4. Urotomografía en corte sagital, fase venosa. Se observa dilatación pielocaliceal bilateral, predominante la izquierda.
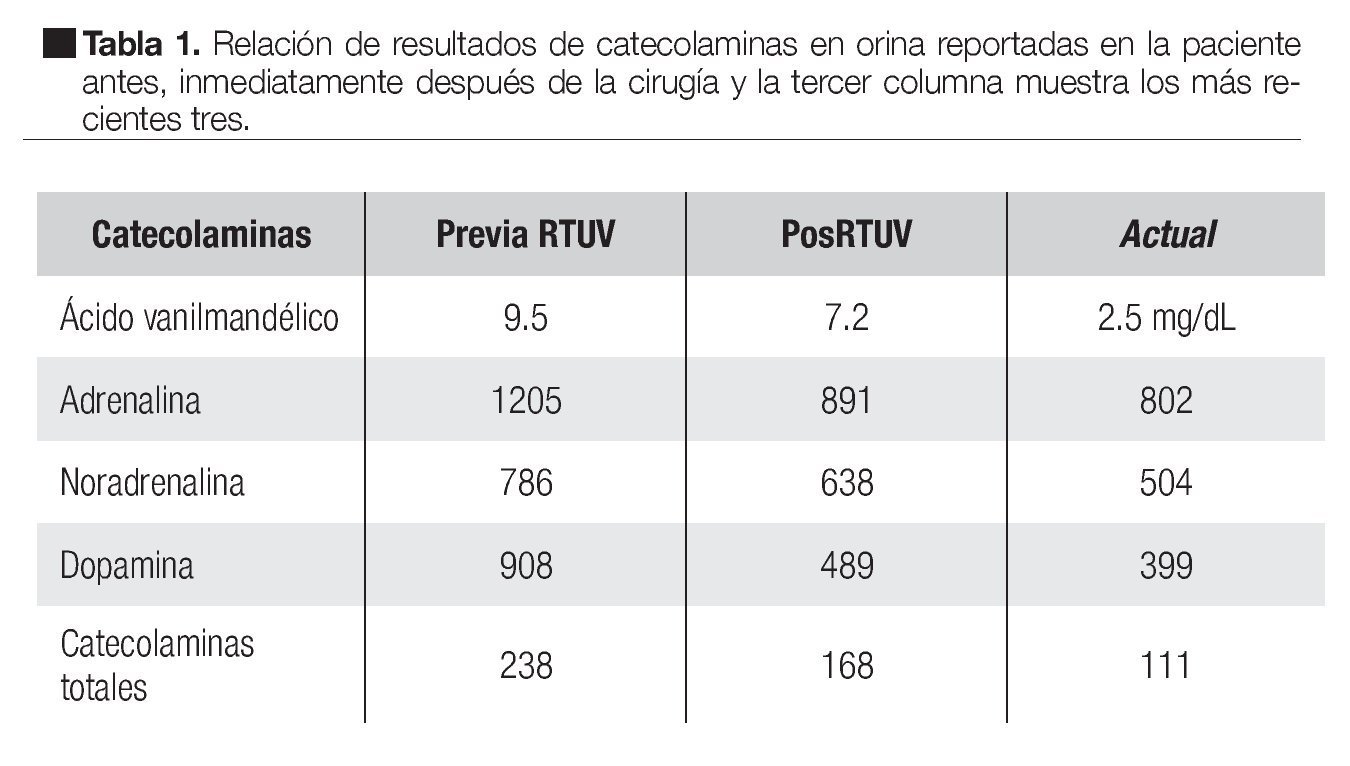
Ante el cuadro clínico de probable feocromocitoma sin imágenes sugestivas de masas suprarrenales, se sospechó probable feocromocitoma extra-adrenal, siendo el sitio de principal sospecha de origen vesical por la masa antes descrita. Se solicitaron catecolaminas en orina informándose dopamina 908 ug/24 horas, adrenalina 1205 ug/24 horas, noradrenalina 786 ug/24 horas, ácido vanililmandelico 9.5 mg/dL.
Ante la contraindicación de biopsia de este tipo de lesiones se decidió tratamiento mediante resección transuretral del tumor vesical, en cuyo informe histopatológico se señaló paraganglioma que infiltra la muscular propia de la vejiga T2a N0 M0. Resultado de inmunohistoquímica: CK7 negativa, CK 20 negativa, CK8 negativa, cromografina positiva, sinaptofisina positivo.
Desde su tratamiento se ha llevado a cistoscopias de control a los tres, seis y 12 meses de pos-operada sin presentar recurrencia tumoral (Tabla 1) además de controles de catecolaminas en orina en descenso desde su diagnóstico.
¿ DISCUSIÓN
El paraganglioma vesical es una entidad dentro de las tumoraciones vesicales no uroteliales, la triada clásica del cuadro clínico es hematuria macroscópica silente, presente en 60% de los casos reportados, hipertensión paroxística y "ataques miccionales" que consisten en cefalea, taquicardia, diaforesis, visión borrosa, entre otros durante o después del vaciamiento vesical. Aunque datos recientes han señalado una herencia genética en 25% a 30% (VHL, NF1), en otras series no se ha encontrado correlación.
Mediante la cistoscopia podemos observar tumores amarillentos que nos obligan a sospechar esta entidad. Dado que el comportamiento biológico de estas lesiones no se puede predecir en base a los hallazgos histológicos (como necrosis, invasión angiolinfática, alto índice mitótico, ausencia de cuerpos hialinos, alteraciones en p53) y a que su comportamiento maligno se ha documentado en 5% a 19% de los paragangliomas, además de ser un tumor quimio y radio-resistente; se recomienda el tratamiento mediante cistectomía parcial;9 sin embargo, en series recientes se ha documentado que dicho tratamiento es el recomendado preferente para lesiones malignas ya conocidas (metastásico).
Con respecto a su seguimiento, Lenders y colaboradores10 han señalado mayor sensibilidad y especificidad en metanefrinas plasmáticas que en las urinarias. La cistoscopia de control se debe realizar en forma regular, así como los estudios de imagen. No hay consenso sobre la frecuencia de las revisiones de control; sin embargo, varios autores han recomendado el seguimiento anual si se encuentran asintomáticos.
Nuestra paciente se encuentra con buena evolución hasta el momento con indicadores negativos de malignidad, sin embargo debido al tratamiento conservador hemos decido mantenerla en control semestral.
¿ CONCLUSIONES
La cistoscopía de control se debe realizar en forma regular, así como los estudios de imagen. No hay consenso sobre la frecuencia de las revisiones de control, sin embargo, varios autores han recomendado el seguimiento anual si se encuentran asintomáticos.
Nuestra paciente se encuentra con buena evolución hasta el momento con indicadores negativos de malignidad, sin embargo debido al tratamiento conservador hemos decido mantenerla en control semestral.
Correspondencia: Dra. Grisel Hernández Martínez.
Blvd. Manuel Avila Camacho SN Lomas de Sotelo Av. Industria Militar y General Cabral, Del. Miguel Hidalgo, C.P. 11200. México, Distrito Federal.
Teléfono: 01 5557 3100, Ext.: 1535.
Correo electrónico:lesirg@gmail.com