


INTRODUCCIÓN
La incidencia de tumores adrenales es baja en la población general: 1 por cada 1.7 millones de habitantes. Representa 0.02% de los tumores y 0.2% de las muertes por cáncer. En el año 2003 se presentaron en México 23 casos nuevos en el plano nacional (0.03% de los cánceres) con una mortalidad de 0.12% de los casos acumulados. En 1996, los casos notificados en el Hospital General Dr. Manuel Gea González fueron cuatro en ocho años. Las manifestaciones clínicas son diversas e incluyen alteraciones hormonales y fisiológicas variadas o algunas relaciones con síndromes genéticos diversos. En la categoría de "incidentalomas" se incluye a los individuos que se han mantenido asintomáticos y cuyo diagnóstico ha sido incidental, al utilizar un método imagenológico. Su frecuencia es elevada, dado que varía de 0.3% a 5 % de todos los pacientes sometidos a estudios imagenológicos abdominales. Si se excluye a los pacientes que tienen lesiones malignas primarias extraadrenales concurrentes, la hemorragia suprarrenal, las lesiones adrenales inflamatorias como la tuberculosis y las micosis, la frecuencia de los incidentalomas verdaderos varía de 0.6% a 1.4%.1,2
El diagnóstico diferencial de una tumoración suprarrenal descubierta de manera incidental en un adulto comprende los siguientes: adenoma cortical, carcinoma adrenocortical, feocromocitoma, hemorragia o fibroma organizado, mielolipoma (tumor producido por tejido adiposo adulto, bien diferenciado, mezclado con elementos hematopoyéticos de las tres series), adenolipoma y metástasis. Por otro lado, los padecimientos que son susceptibles de tratamiento quirúrgico electivo de adrenalectomía son hiperaldosteronismo primario, hiperadrenocortisolismo, además de los casos anteriores.3 En la actualidad, el abordaje de elección es la vía laparoscópica y es el tratamiento de elección, siempre que se disponga de todos los recursos tecnológicos y personal entrenado en este tipo de procedimientos.4
OBJETIVO
Presentar la experiencia de 20 años en el manejo quirúrgico de la patología adrenal en el Servicio de Urología del Hospital General Dr. Manuel Gea González.
MATERIAL Y MÉTODOS
Se identificaron los registros quirúrgicos y los expedientes clínicos de los casos con lesiones suprarrenales tratados en el servicio de urología entre 1988 y 2008. Se analizaron 12 casos, de los cuales se excluyeron tres porque no contaban con expediente clínico completo. El resto se integró con nueve casos, de los que se registraron los hallazgos histopatológicos. Se revisaron los datos sociodemográficos, la forma de presentación clínica y la actividad metabólica, los datos de laboratorio e imagen que apoyaron el diagnóstico y la técnica quirúrgica realizada para el tratamiento, complicaciones y pronóstico de seguimiento.
RESULTADOS
Se presentaron nueve casos de tumores suprarrenales para tratamiento quirúrgico, cuatro correspondientes a varones y cinco a mujeres, con edades fluctuantes entre 35 y 63 años en aquéllos y 30 y 65 años en éstas.
De acuerdo con el lado afectado, se presentaron siete tumores en las glándulas del lado derecho y dos en las glándulas del izquierdo. En relación con sus características clínicas y metabólicas, se subdividieron los casos en tumores suprarrenales funcionales (n = 5) y no funcionales (n = 4).
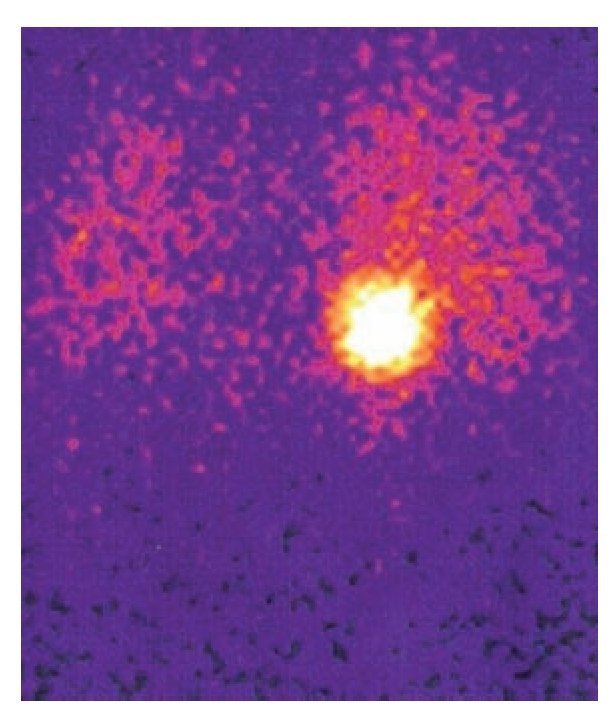
Dentro de las masas funcionales se identificaron cuatro casos de feocromocitoma, que afectó a dos varones y dos mujeres. Los cuatro fueron tumores del lado derecho y en todos se presentó hipertensión arterial como manifestación clínica constante y refractaria al tratamiento médico de dos antihipertensivos; en tres, las crisis adrenérgicas fueron constantes y ocasionaron sudación profusa, mareos y sensación de nerviosismo con cambios de coloración facial. Un caso se diagnosticó por hallazgo incidental secundario a pancreatitis aguda y cuya única manifestación fue la hipertensión arterial refractaria. En los cuatro casos se determinaron metanefrinas en orina y acido vanililmandélico, los cuales fueron positivos para el diagnóstico. Sólo en dos de ellos se obtuvo gammagrama con metayodo-bencilguanidina (MIBG), que fue positivo para demostrar la lesión adrenal, y no se encontró tejido cromafín en sitios extraadrenales. Todos los casos recibieron tratamiento médico preoperatorio con bloqueadores alfa y beta, además de carga hídrica antes de la operación; tres pacientes se sometieron a una adrenalectomía derecha por abordaje abierto y uno por abordaje laparoscópico. En dos se presentaron crisis adrenérgicas moderadas en el transoperatorio, que se resolvieron sin complicaciones. El tamaño tumoral osciló entre 4 y 8 cm, con una media de 6.5 cm. La evolución posoperatoria de los cuatro casos fue buena y no se presentó un comportamiento maligno en ninguno de los casos. El tiempo de seguimiento fue de tres meses a 12 años, sin mortalidad.
Se informó de un tumor de corteza adrenal en una mujer de 52 años con hipertensión arterial y síndrome de Cushing clínico; se obtuvo un perfil funcional hormonal y se confirmó el diagnóstico por las cifras de cortisol; dadas las características clínicas y el tamaño tumoral (9 x 7 cm), se sospechó malignidad, se le sometió a resección adrenal izquierda por vía laparoscópica y el diagnóstico histopatológico estableció adenoma suprarrenal funcional, corroborado con pruebas de inmunohistoquímica. Tiene hasta el momento un seguimiento de cuatro años después de la intervención sin complicaciones metabólicas.
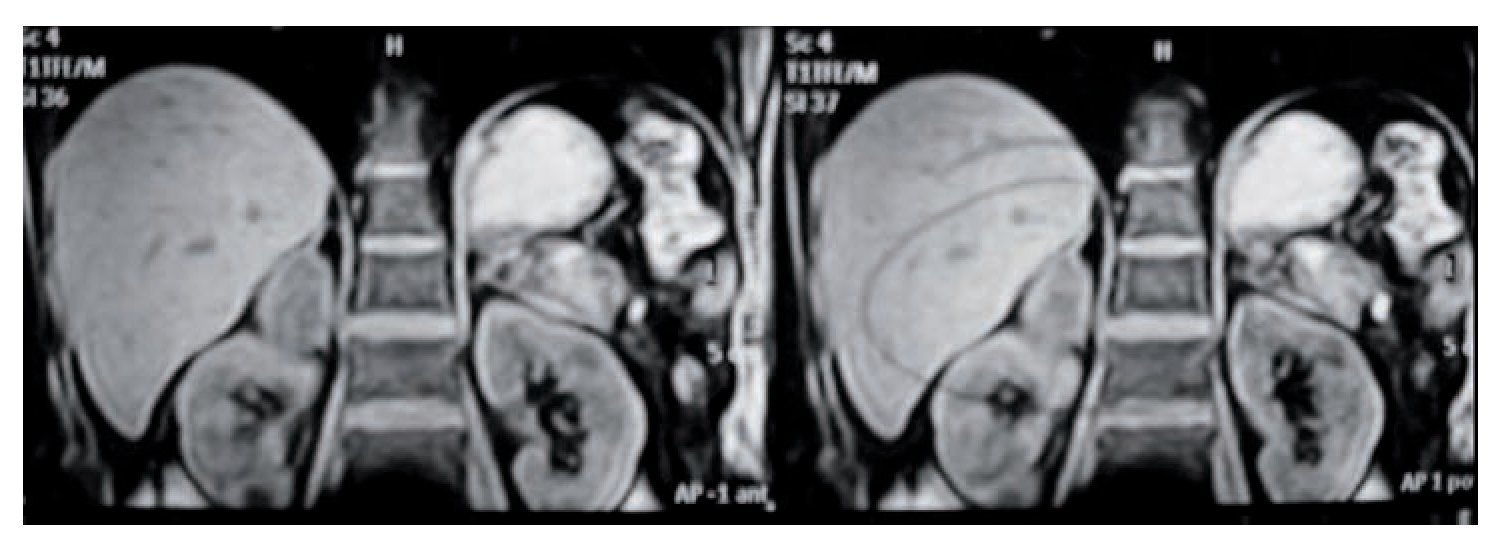
De las masas adrenales no funcionales (4), se presentaron dos incidentalomas en dos pacientes masculinos de 59 y 60 años de edad. Ambos se diagnosticaron en estudios de ultrasonido abdominal y se determinó la masa adrenal con estudios de extensión como TAC y resonancia magnética (Imágenes 1 y 2). Los hallazgos se relacionaban con la glándula suprarrenal derecha. En uno de ellos se encontró además una imagen de quiste complejo de Bosniack tres en el riñón ipsolateral, por lo que se sometió a exploración quirúrgica con resección de la masa quística renal y adrenalectomía; se encontró un quiste simple renal y un mielolipoma de 4 x 4 cm, sin complicaciones transoperatorias, con un tiempo de seguimiento de seis meses asintomático. El segundo caso se sometió a adrenalectomía abierta con informe de mielolipoma de 8 x 8 cm, sin complicaciones y con un seguimiento de 12 años asintomático. En un sujeto de 30 años se encontró una masa abdominal gigante, que dio motivo a la búsqueda de atención médica; la TAC delineó tumor quístico dependiente de glándula suprarrenal derecha, para lo cual se programó una operación con abordaje toracoabdominal, sin complicaciones y conservación asintomática a 12 años de seguimiento (Imagen 3).
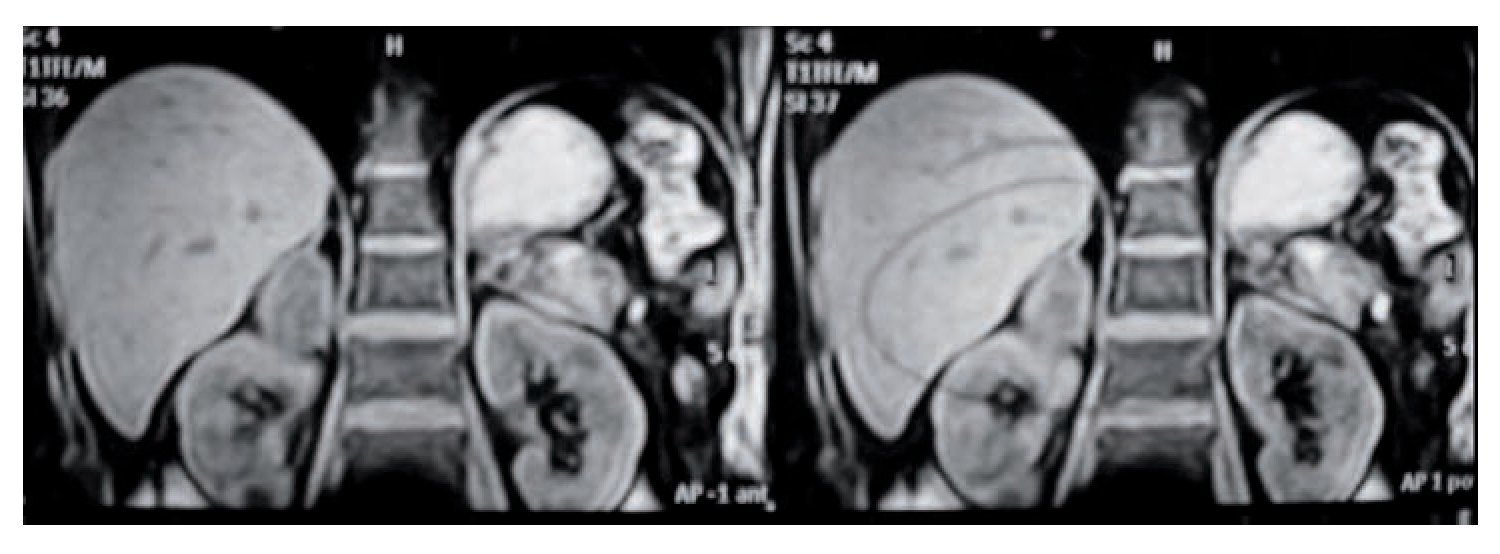
Imagen 1. Feocromocitoma derecho por resonancia magnética.
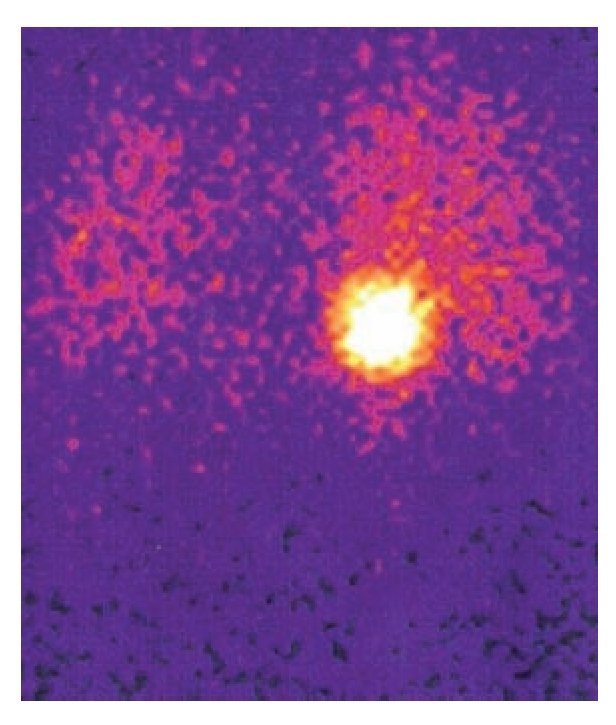
Imagen 2. Gammagrama con MIBG que muestra feocromocitoma derecho sin tejido cromafín en sitios extraadrenales.
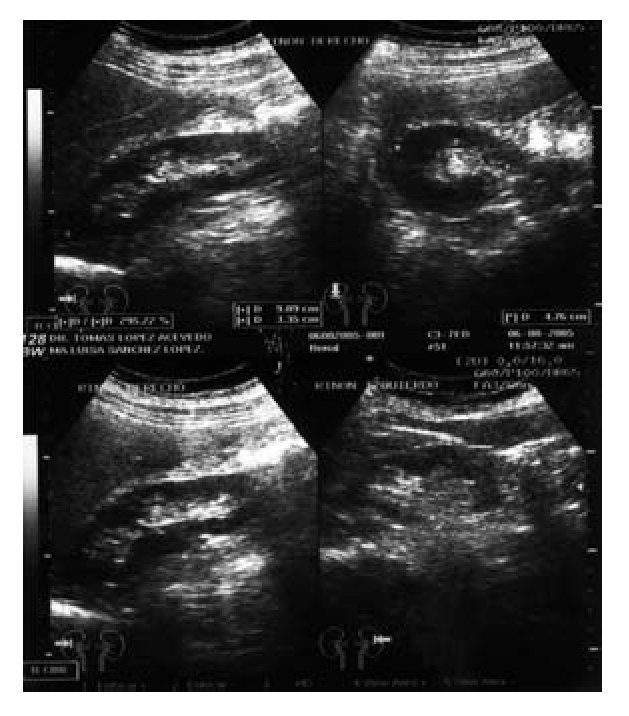
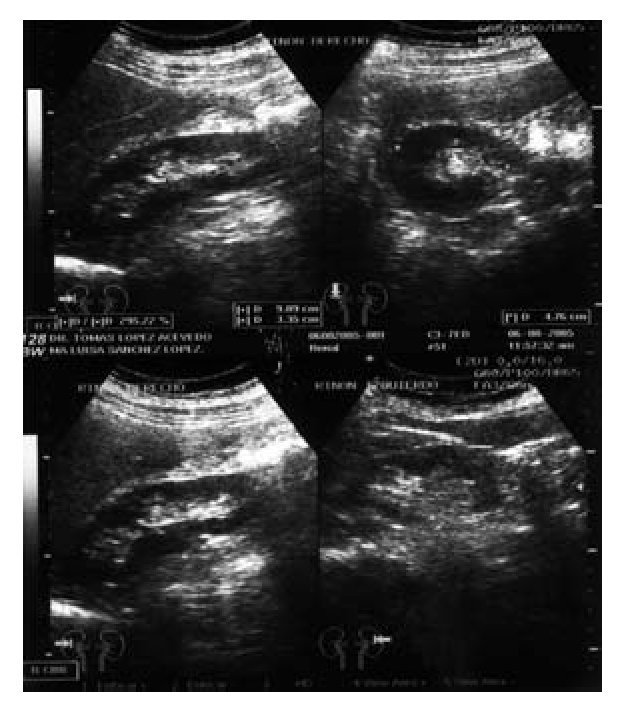
Imagen 3. Ultrasonido renal con masa hipoecoica de 3.71 cm, medial al polo superior del riñón izquierdo.
Por último, se reconoció un caso de leiomiosarcoma adrenal primario en una paciente femenina de 65 años de edad que presentó de manera inicial hipertensión refractaria a tratamiento médico (clasificada como hipertensión renovascular con estenosis de la arteria renal del riñón izquierdo). En los estudios de extensión se encontró una masa adrenal con afectación del retroperitoneo, evidente en los estudios de TAC y resonancia magnética. Las determinaciones funcionales de catecolaminas y hormonales fueron negativas, por lo que se sometió a la intervención, en la que se identificó una tumoración de 5 x 5 cm, que se resecó en su totalidad; sin embargo, el informe posoperatorio confirmó un leiomiosarcoma adrenal primario con bordes positivos. La paciente decidió no someterse a un nuevo procedimiento quirúrgico y se mantuvo en vigilancia, ha tenido un seguimiento de tres años tras la operación y en ese lapso ha requerido manejo endovascular la arteria renal izquierda por hipertensión renovascular, con mejoría de ésta. No se encontraron datos de actividad tumoral. El tratamiento quirúrgico se efectuó por vía abierta sin complicaciones.
DISCUSIÓN
Los tumores de la glándula suprarrenal son poco frecuentes y su baja incidencia determina la existencia de gran variedad de tipos histológicos y formas clínicas de presentación.
El feocromocitoma es un tumor raro de células cromafines que se origina casi siempre en la médula de la glándula suprarrenal. Se calcula que en Estados Unidos se presentan cada año 800 casos.5 La mayor incidencia se observa entre los 30 y 50 años de edad. Una proporción de 10% corresponde a masas bilaterales y es más común el feocromocitoma familiar, que se presenta con los síndromes familiares de neoplasia endocrina múltiple (MEN, tipos 2A y 2B). En los casos atendidos en la institución de los autores se observó un predomino de la glándula adrenal derecha sin distinción de género y sin alteraciones adjuntas. En pacientes con síndromes MEN tipo 2, el riesgo de desarrollar un tumor contralateral después de adrenalectomía unilateral se aproxima a 50%. Otros síndromes relacionados con feocromocitoma son la neurofibromatosis, la enfermedad de von Hippel-Lindau, el hemangioblastoma cerebeloso, el síndrome de Sturge-Weber y la esclerosis tuberosa.6-8 El feocromocitoma extraadrenal o paraganglioma se presenta en 10% a 15% de los casos y puede originarse de cualquier tejido cromafín extraadrenal, frecuentemente en los ganglios simpáticos. El diagnóstico se establece con la determinación de catecolaminas libres (norepinefrina y epinefrina) o metabolitos de catecolaminas (ácido vanililmandélico y metanefrinas totales) en orina de 24 horas. La determinación de catecolaminas plasmáticas también puede tener valor en el diagnóstico, aunque muestra una sensibilidad y una especificidad limitadas.3,4,9 Los estudios iniciales deben incluir una radiografía del tórax y una tomografía computarizada (TAC) del abdomen. El gammagrama con metayodobencil-guanidina (MIBG) es un estudio útil como método para la localización tumoral y descartar tejido cromafín en otros planos extraadrenales.10-13 El tratamiento adecuado es quirúrgico, con preparación preoperatoria, bloqueo adrenérgico alfa (fentolamina, prazosina, terazosina, doxazosina, fenoxibenzamina) para controlar la presión arterial y la expansión del volumen extracelular; de esta forma es posible reducir las posibles variaciones tensionales o las arritmias durante la anestesia o durante la intervención quirúrgica.13-16 El tratamiento se instituye dos a tres semanas antes del acto operatorio. Con posterioridad pueden indicarse bloqueadores beta (propanolol, metoprolol, atenolol, esmolol) o un bloqueador adrenérgico alfa y beta tipo labetalol. En los casos de este protocolo se iniciaron dos y tres semanas anteriores a la intervención, con lo que se obtuvo una adecuada respuesta en dos casos, ya que en dos más se presentaron crisis adrenérgicas transoperatorias moderadas, sin consecuencias posteriores.5,7,12
Las neoplasias primarias del mesénquima adrenal pueden ser benignas, entre ellas los hemangiomas cavernosos, hemangiomas capilares, lipomas, neurofibromas, tumores adenomatoides y leiomiomas de venas adrenales, o malignas como los sarcomas. Entre los sarcomas originados en la glándula suprarrenal se han descrito los siguientes: angiosarcomas, leiomiosarcomas y tumores malignos de la vaina nerviosa periférica. Los leiomiosarcomas adrenales son infrecuentes y existen pocos casos informados en las publicaciones internacionales, tres de ellos vinculados con el síndrome de inmunodeficiencia adquirida. En la bibliografía revisada no se encontró ningún otro factor etiológico común entre los casos. Por lo anterior, se trata de un caso excepcional y su tratamiento debe ser radical, ya que no responden a la quimioterapia o radioterapia.16,17
Los quistes suprarrenales son anomalías poco habituales y sólo se encuentran en informes de casos y muy escasas revisiones de series. En términos de su evolución natural, tienden a mantenerse asintomáticos hasta que los síntomas aparecen por el crecimiento. La manifestación clínica más común es el dolor abdominal. Su tamaño es variable al momento del diagnóstico y el tratamiento es quirúrgico, sea por abordaje abierto o por vía laparoscópica. Por lo regular, sus dimensiones oscilan entre 5 y 10 cm, con un promedio de 9.6 cm, aunque se han notificado malformaciones mayores de 50 cm. El caso presentado fue de 20 cm y requirió un abordaje toracoabdominal. Son unilaterales en el 80% de los casos y apenas un 7% corresponde a masas malignas o potencialmente malignas y un 0.2 % tiene un origen metastásico.
El mielolipoma adrenal constituye un tumor benigno raro, hormonalmente inactivo, compuesto por tejido adiposo maduro y tejido hematopoyético que simula la médula ósea. Casi siempre es asintomático y se diagnostica de forma incidental. Representa sólo el 11% de los tumores adrenales y su tratamiento es quirúrgico, abierto o laparoscópico. En algunos casos puede experimentar rotura espontánea, lo que ocasiona hemorragia grave, hipertensión y nefroangioesclerosis.7,10 En los casos presentados se trata de lesiones que se encontraron incidentalmente con una resolución quirúrgica sin complicaciones.
CONCLUSIONES
La incidencia de la patología adrenal es baja en la población atendida por los autores; la presencia de nueve casos en 20 años en una institución de consulta general abierta hace evidente que su diagnóstico tiene un bajísimo índice de sospecha y frecuentemente se encuentran de forma incidental. El tratamiento de esta anomalía no es exclusivo del urólogo y casi siempre se atiende en los servicios de cirugía general y cirugía endocrina, por lo que es difícil encontrar grandes series de casos. Los tumores cromafines son los más frecuentes por tipo tumoral en la institución, pero el número de casos es bajo. El resto de los tumores tiene una histología variable y su evolución establece con frecuencia el diagnóstico. El abordaje debe ser multidisciplinario para precisar un adecuado diagnóstico, manejo y pronóstico a largo plazo y el urólogo debe estar familiarizado con este tipo de padecimientos y las técnicas quirúrgicas actuales para su resolución adecuada.
Correspondencia: Dr. Mauricio Cantellano Orozco.
División de Urología, Hospital General Dr. Manuel Gea González, Secretaría de Salud, México D. F.
Calzada de Tlalpan 4800. Col. Toriello Guerra. Delegación Tlalpan. C.P. 14000.
Teléfono: 4000-3000, ext. 3044, fax 4000-3044.
Correo electrónico:mcantellanomd@yahoo.com.mx