


El cáncer renal sigue siendo una enfermedad quirúrgica. Los tumores renales cromófobos tienen mejor pronóstico que otras variantes histológicas y suelen presentarse de manera esporádica o asociados a síndromes familiares. La nefrectomía parcial está indicada cuando se prevé la pérdida total de la función renal.
Se presenta paciente femenino de 45 años de edad, con antecedente de nefrectomía izquierda por donación, sin vigilancia de riñón único. Acude por masa abdominal y prurito de predominio nocturno. La tomografía abdominal mostró tumoraciones en riñón derecho y útero. El laboratorio reportó función renal normal, marcadores tumorales para cáncer de ovario negativos y función hepática alterada. Se realizó histerectomía y heminefrectomía derecha con isquemia caliente de 19 minutos, el cierre del parénquima renal se cubrió con un parche de tejido adiposo y se colocó catéter ureteral. La función hepática se normalizó después de la nefrectomía. El estudio histopatológico reportó carcinoma cromófobo de células renales y leiomiomatosis uterina.
Es conveniente un seguimiento periódico a largo plazo de los pacientes con riñón único. No se integró un síndrome de cáncer renal hereditario en esta paciente, sin embargo, el tratamiento debe encaminarse a preservar el mayor número de nefronas independientemente de la estirpe histológica.
Kidney cancer continues to be a surgical disease. Chromophobe type kidney tumors have a better outcome than other histologic variants and usually present sporadically or are associated with familial syndromes. Partial nephrectomy is indicated when the total loss of kidney function is foreseen.
A 45-year-old woman had a past history of left nephrectomy due to kidney donation and she had no surveillance of the remaining kidney. The patient sought medical attention for an abdominal mass and predominantly nocturnal pruritus. An abdominal tomography scan revealed tumors in the right kidney and uterus. Laboratory tests reported normal kidney function, negative tumor markers for ovarian cancer, and altered liver function. Hysterectomy and right heminephrectomy with 19 minutes of warm ischemia were performed. The kidney parenchyma was closed with an adipose tissue patch and a ureteral stent was placed. Liver function normalized after the nephrectomy. The histopathologic study reported chromophobe renal cell carcinoma and uterine leiomyomatosis.
A long-term follow-up period is recommended for patients with a solitary kidney. This patient did not present with a hereditary kidney cancer syndrome, but nevertheless, treatment should strive to spare the greatest number of nephrons, regardless of the histologic strain.
Introducción
El carcinoma de células claras representa más del 80% de los tumores renales1, seguido por el carcinoma papilar y el carcinoma de células cromófobas2-4. La mayoría de los tumores renales cromófobos son esporádicos, pero ocasionalmente se encuentran asociados con el síndrome de Birt-Hogg-Dubé5 y se considera que tienen mejor pronóstico que el carcinoma de células claras1,2,5. El diagnóstico diferencial de los tumores cromófobos no es fácil, por lo que puede requerirse el uso de auxiliares diagnósticos como estudios de inmunohistoquímica6.
Al igual que los tumores de próstata, colon o mama, el cáncer renal puede presentarse de forma esporádica o de forma hereditaria. Se han descrito varios tipos hereditarios de cáncer renal7. En 2001, se describió un síndrome de cáncer renal familiar en el cual los pacientes desarrollan leiomiomas cutáneos y uterinos, y carcinoma de células renales papilar de tipo 24,7-9.
El síndrome de Stauffer es un síndrome paraneoplásico caracterizado por disfunción hepática no metastásica que se normaliza después de la nefrectomía, se ha comunicado en el 3%-20% de los pacientes con cáncer renal.
El tratamiento de los tumores renales es primordialmente quirúrgico, el objetivo es escindir todo el tumor con un margen quirúrgico suficiente4. Cuando el paciente tiene riesgo elevado de necesidad final de diálisis o cuando la nefrectomía implica dejar al paciente anéfrico, está indicada la nefrectomía parcial.
No hay datos convincentes que indiquen que los donantes vivos tengan un mayor riesgo a largo plazo de desarrollar nefropatía crónica debido a la donación renal. No obstante, se recomienda una evaluación de seguimiento periódica a largo plazo de los donantes. De ello puede encargarse el médico personal del donante10.
El objetivo de este estudio es mostrar la conducta terapéutica adoptada para tratar a una paciente con cáncer renal en un riñón solitario y su completa consonancia con lo reportado en la literatura médica. No hubo datos clínicos o paraclínicos suficientes para integrar un síndrome de cáncer renal hereditario, se trata más bien de una paciente con tumor renal cromófobo esporádico asociado de manera incidental a miomatosis uterina que cursó con síndrome de Stauffer.
Presentación del caso
Femenina de 45 años de edad, cuya madre cursó con cáncer de mama y de riñón sincrónicos. Tiene antecedente de nefrectomía izquierda por donación hace 6 años, 1 gesta, 1 parto. Nunca se ha realizado Papanicolaou. Sin seguimiento o vigilancia por riñón único.
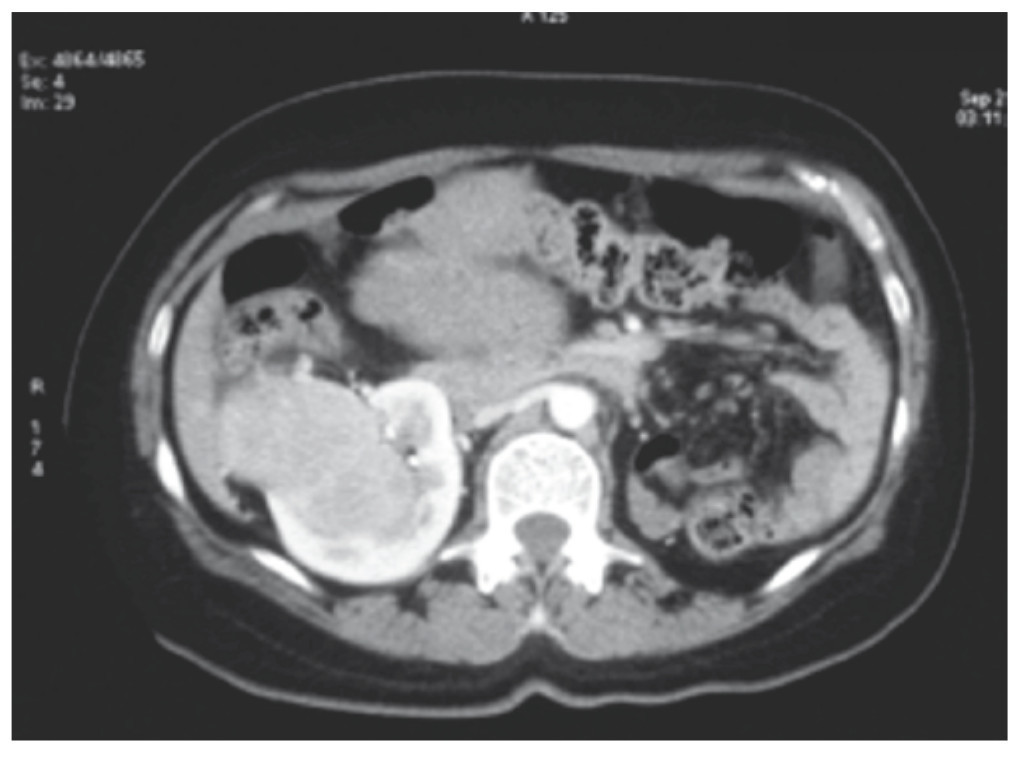
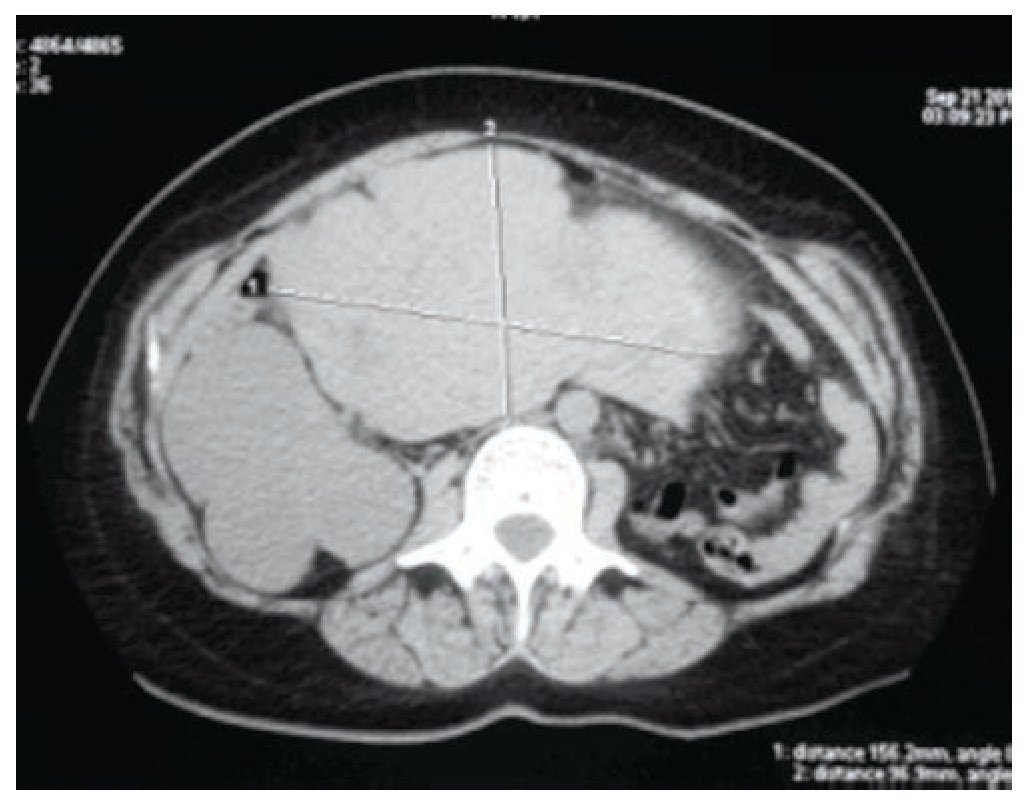
Fue referida a nuestra consulta por hallazgo incidental de tumoración abdominal y renal derecha en ultrasonido abdominal realizado para reanudar la vigilancia de riñón único, además, presentaba desde 2 años previos prurito generalizado de predominio nocturno y masa palpable en abdomen inferior, sin dolor ni síntomas acompañantes. Durante la exploración física se encontró el abdomen con masa palpable en hipogastrio, móvil, no dolorosa; no se apreció ninguna otra alteración. Se realizó tomografía computarizada (TC) de abdomen, en la cual se encontró una tumoración renal derecha estadio T2b de la clasificación TNM y tumoración abdominal dependiente de útero (figs. 1 y 2). La radiografía de tórax se mostró normal. Los exámenes de laboratorio reportaron urea 24 mg/dL, creatinina 0.82 mg/ dL, perfil hepático con bilirrubina total 0.4 mg/dL, fosfatasa alcalina 1,140 U/l, TGO 210 U/l, TGP 185 U/l, GGT 751 U/l, Ca-125 46.25 U/mL, Ca 19-9 7.34 U/mL, antígeno carcinoembrionario 1.16 ng/mL, alfa fetoproteína 3.08 U/mL.
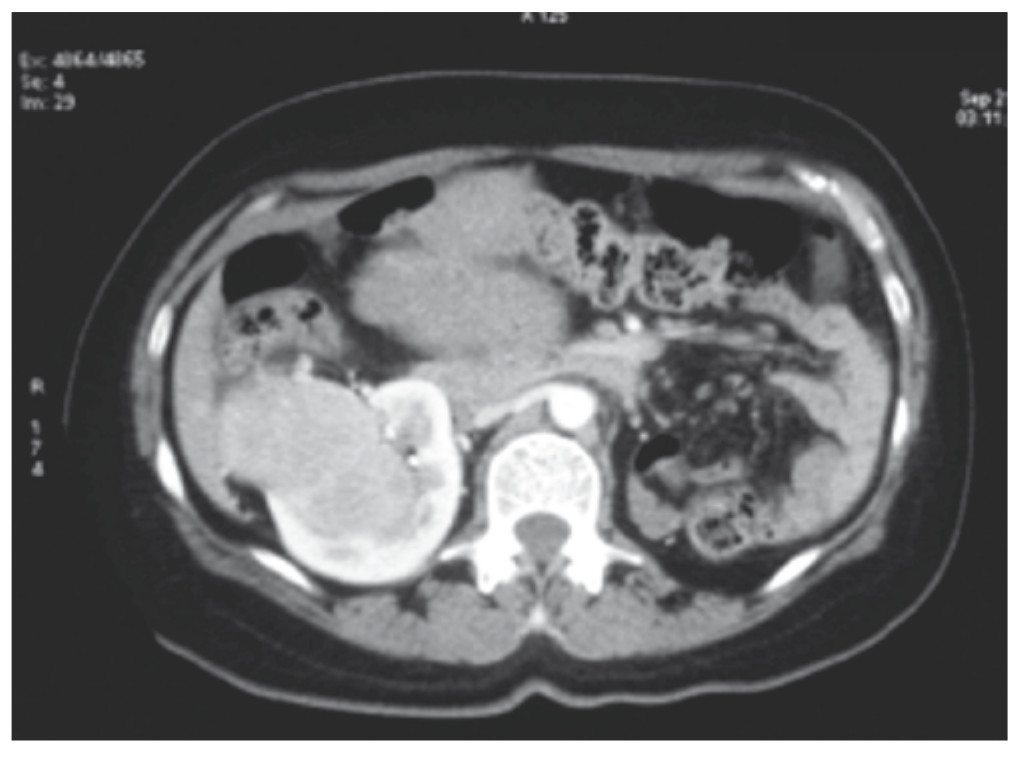
Figura 1 Tomografía axial computarizada que muestra la presencia de una tumoración en el polo inferior del riñón derecho y ausencia de riñón izquierdo. Se aprecia la parte superior de la tumoración uterina.
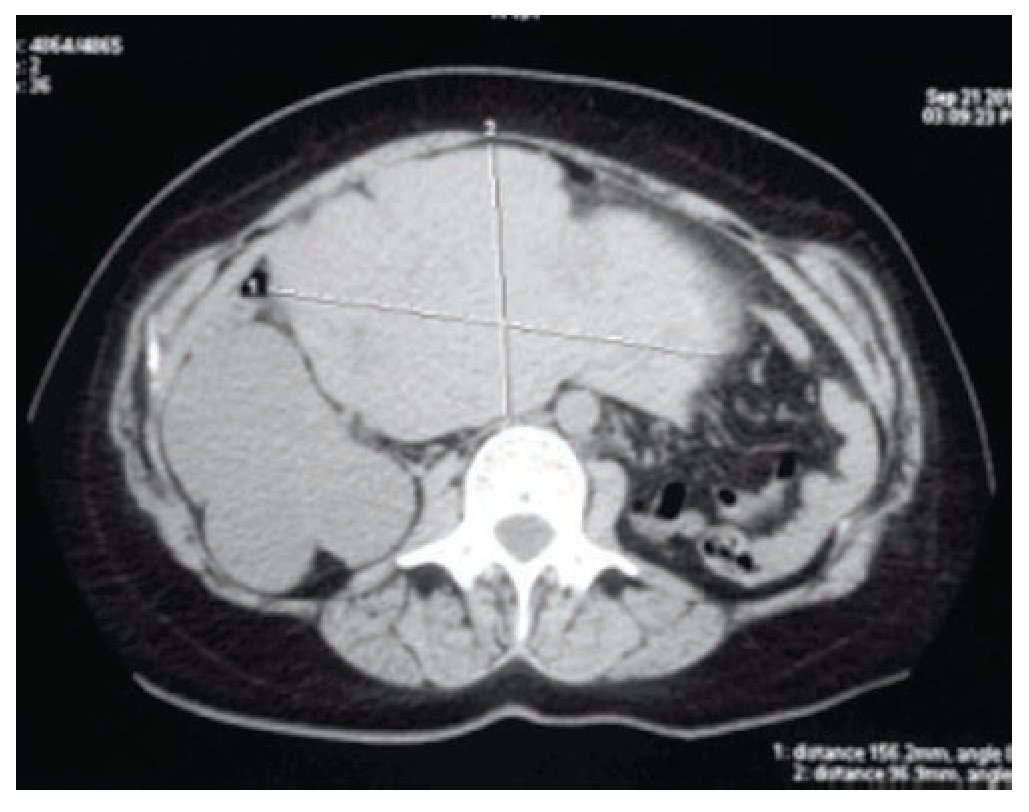
Figura 2 Tomografía axial computarizada que muestra las dimensiones de la tumoración uterina, 156 mm en sentido transversal y 96 mm en sentido anteroposterior.
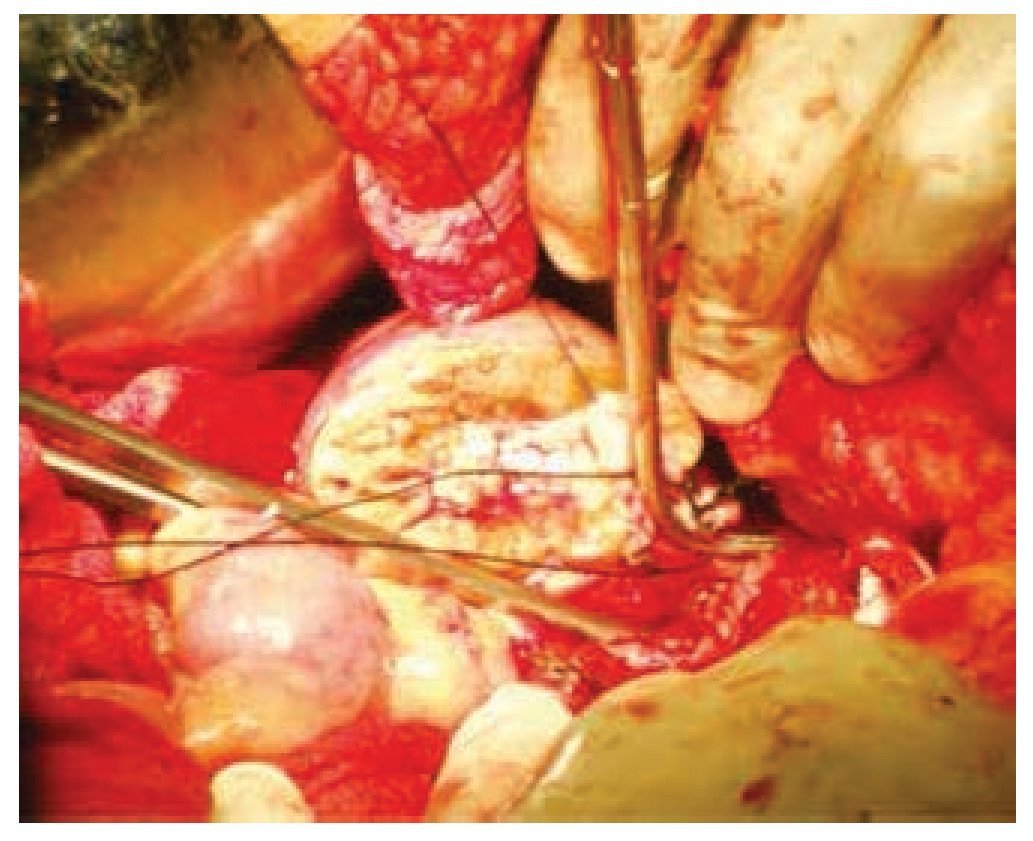
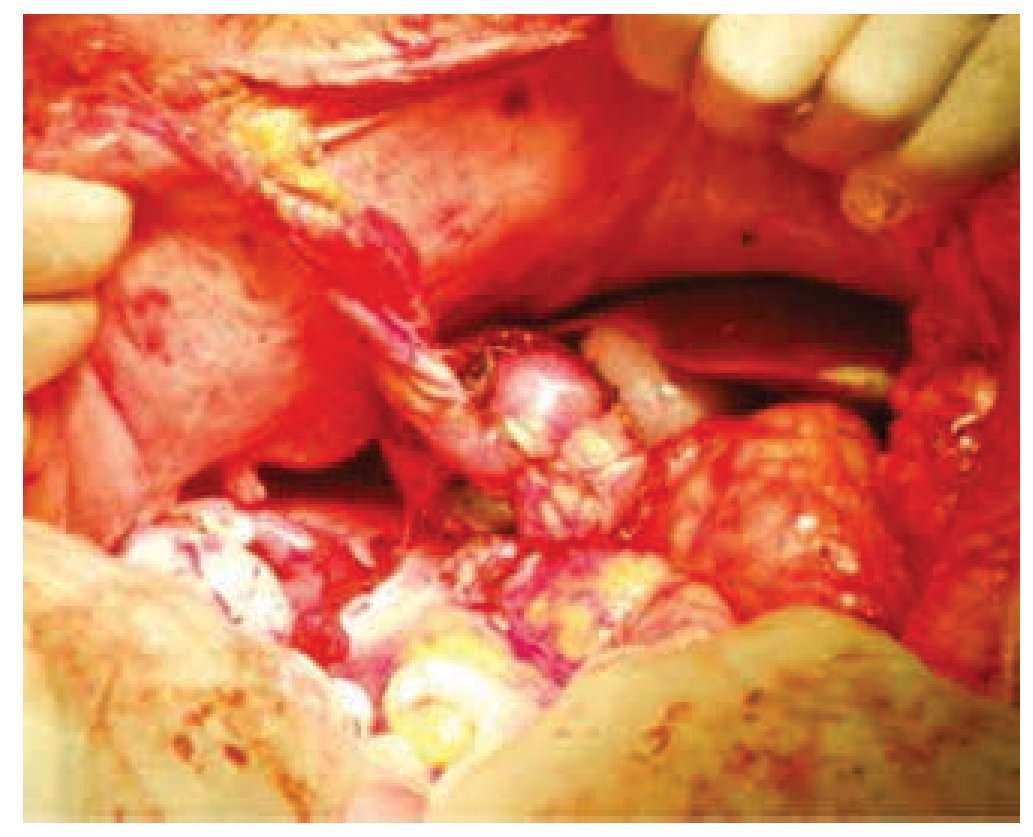
Se realizó laparotomía exploradora, en la cual se encontró una tumoración dependiente de polo inferior de riñón derecho de aproximadamente 10 x 10 x 6 cm, y otra tumoración uterina de superficie irregular y multinodular de aproximadamente 20 x 20 x 9 cm. Se realizó heminefrectomía derecha con isquemia normo térmica de 19 minutos, el cierre del parénquima renal se cubrió con un parche de tejido adiposo pediculado dependiente de ligamento redondo del hígado y se colocó catéter ureteral doble J (figs. 3 y 4); posteriormente se procedió a realizar histerectomía y salpingooforectomía derecha.
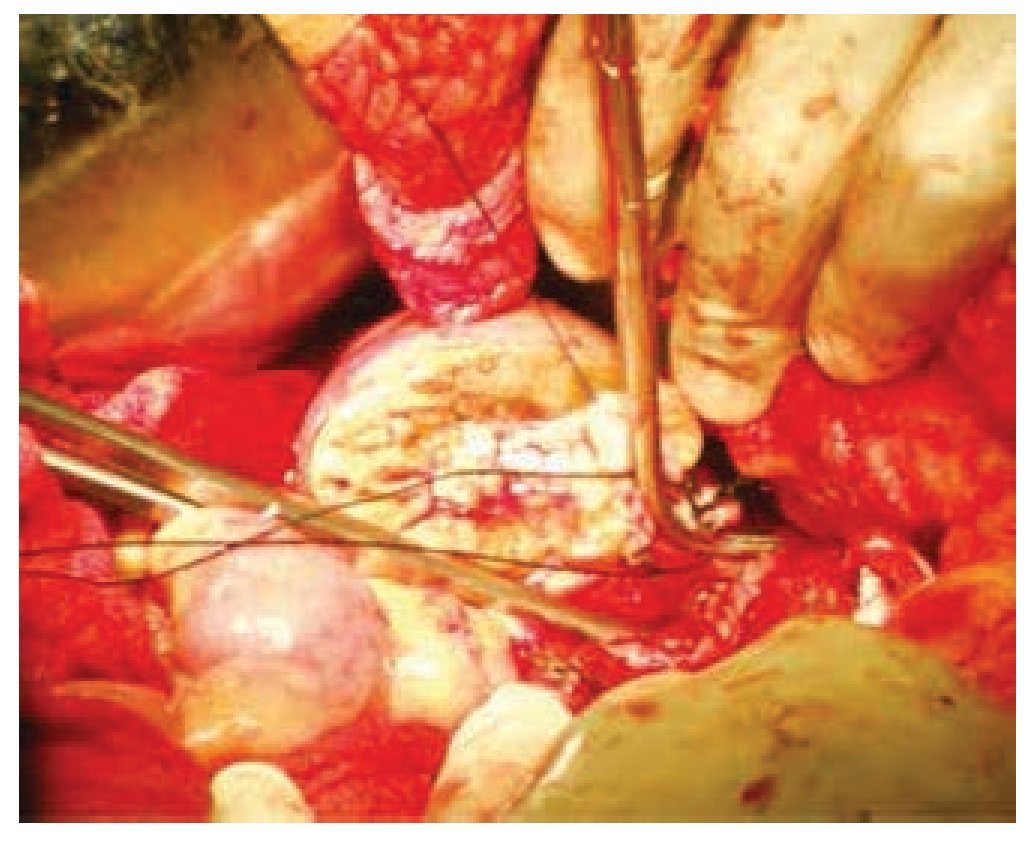
Figura 3 Cierre del sistema colector durante la nefrectomía parcial derecha.
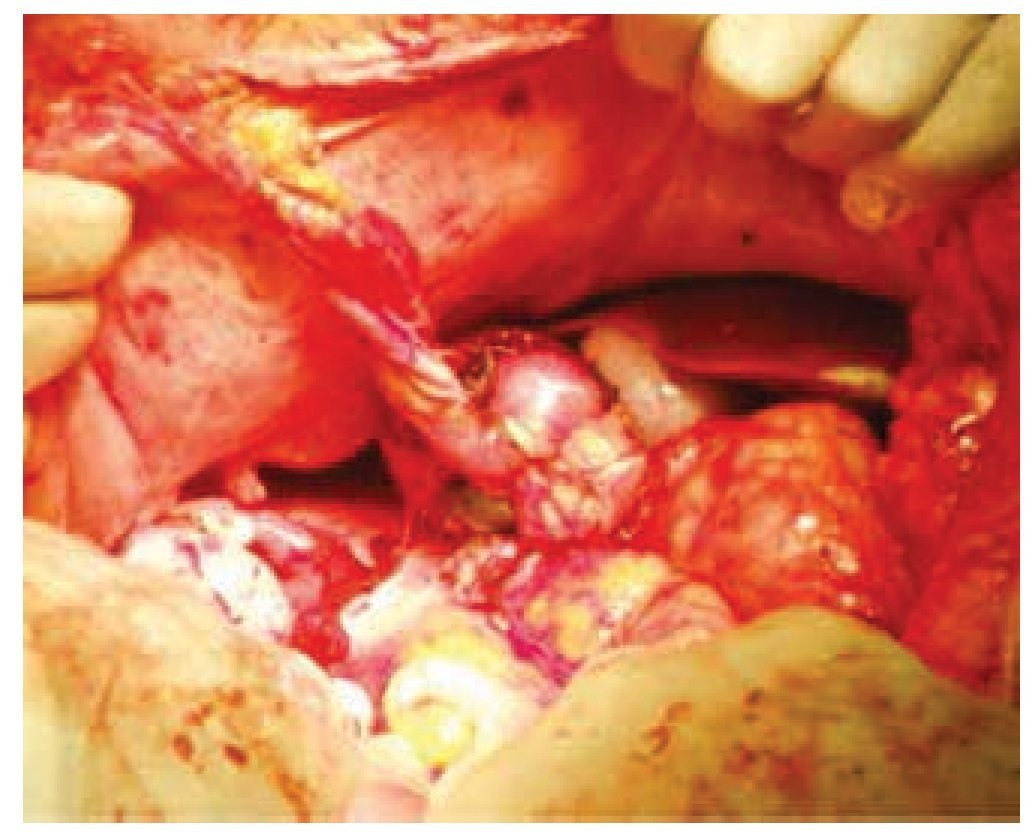
Figura 4 Parche pediculado de tejido adiposo sobre el cierre del parénquima renal.
Durante los primeros días del postoperatorio presentó evolución adecuada. Las pruebas de funcionamiento hepático se normalizaron después de la nefrectomía. A partir del 3° día presentó gasto aumentado por el drenaje, anuria, leucocitosis (22.9 x103/μL con neutrofilia de 93%) y elevación de azoados (creatinina: 3.06 mg/dL, urea: 70 mg/dL), por lo que fue necesario recambiar el catéter doble J previa cistoscopia, cistografía y pielografía ascendente, las cuales descartaron lesión ureteral o vesical. Con esto aumentó la diuresis, se normalizó la cifra leucocitaria y la función renal, y disminuyó la producción del drenaje.
Se dio de alta hospitalaria y el drenaje se retiró a los 7 días. El catéter ureteral se retiró 8 semanas después del evento quirúrgico.
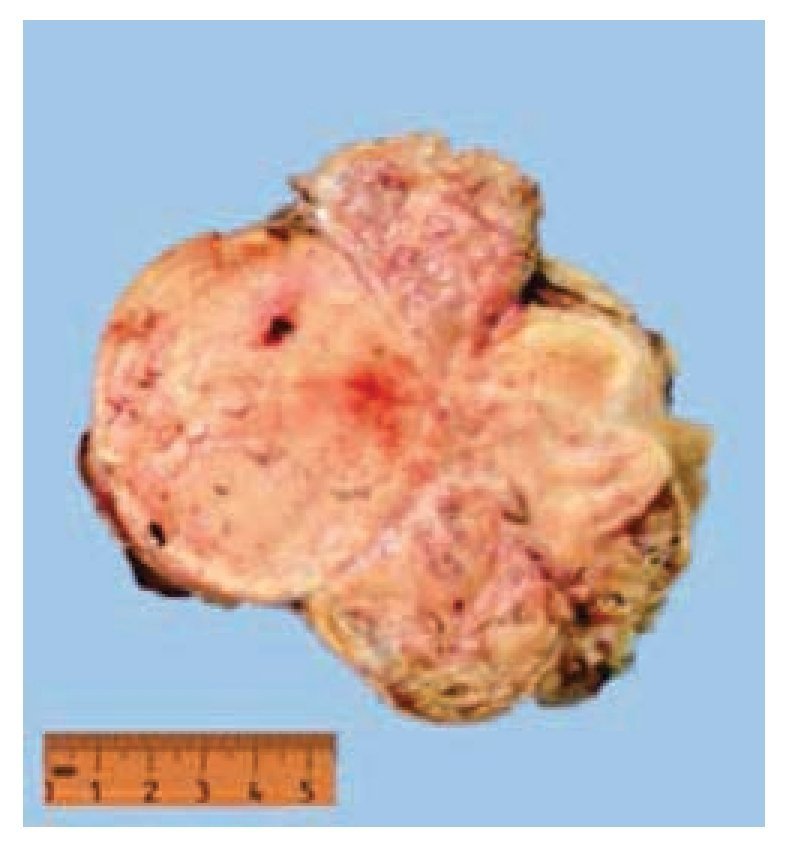
El estudio histopatológico reportó un carcinoma cromófobo de células renales tipo clásico Fuhrman 2 pT2bNxMx, al cual se re realizaron estudios de inmunohistoquímica y resultó positivo para los marcadores CK7 y S100, leiomiomatosis uterina y líquido peritoneal positivo para tumor cromófobo (figs. 5 y 6).
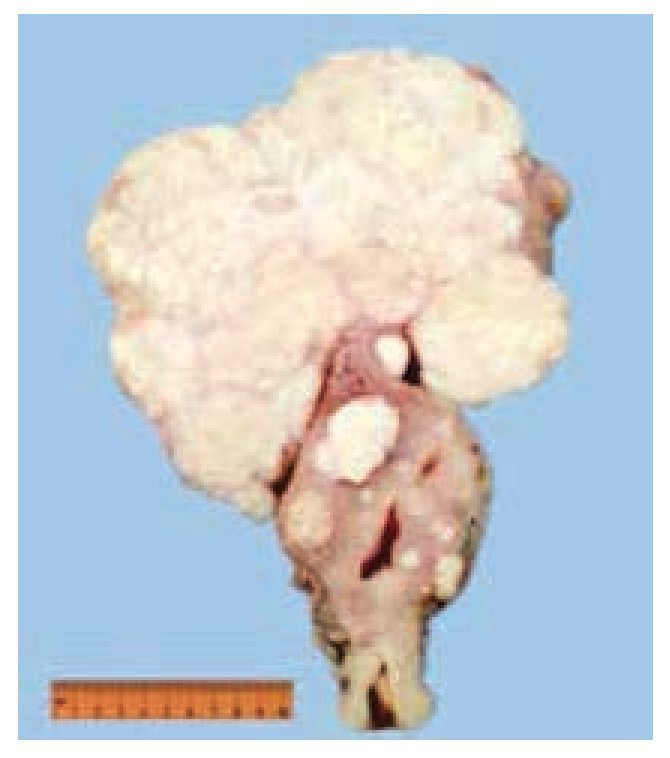
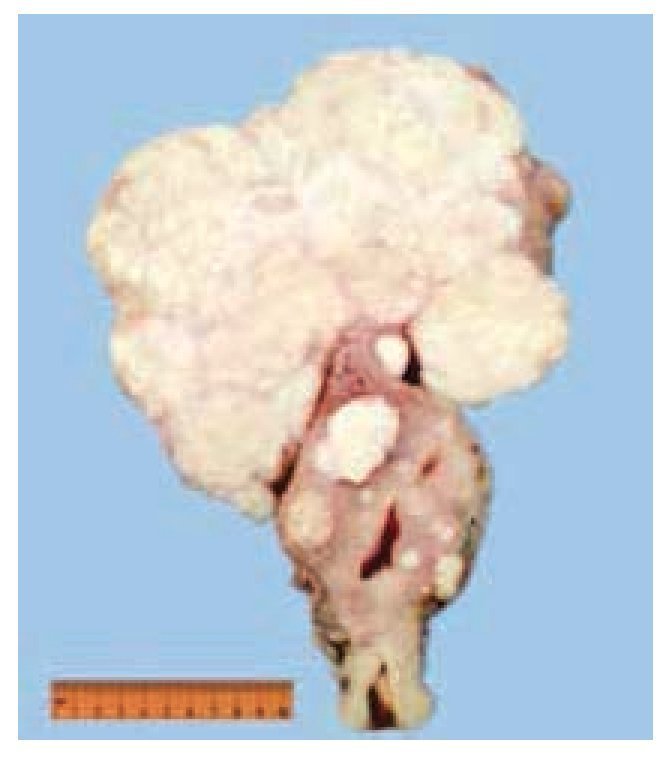
Figura 5 Útero con miomas. Se evidencia la presencia de múltiples tumoraciones intramurales y una gran tumoración subserosa, de superficie homogénea de color café claro y consistencia blanda.
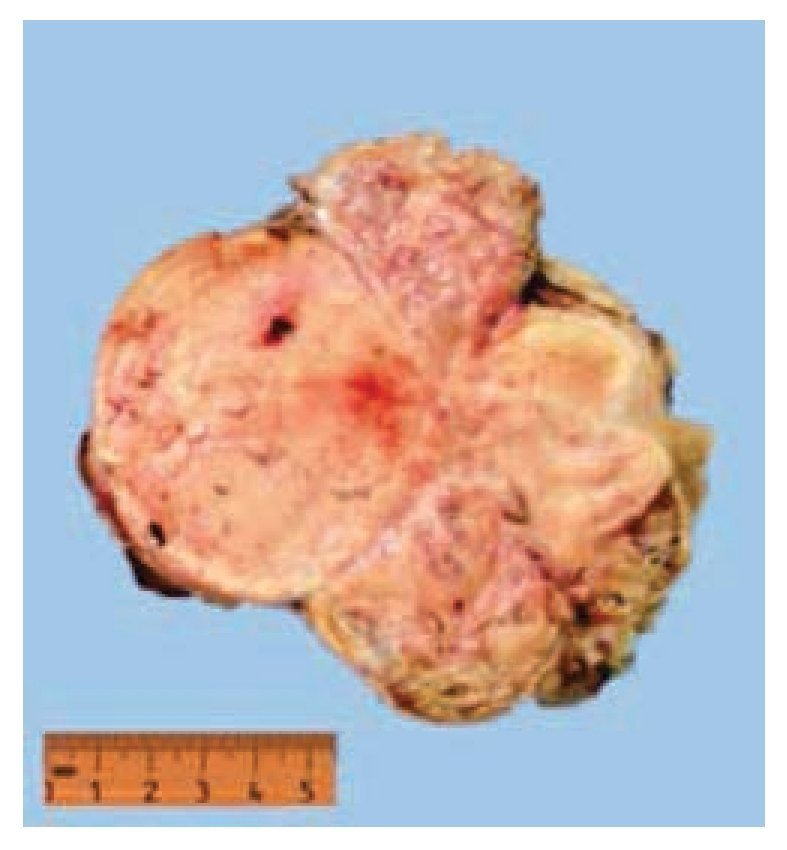
Figura 6 Tumor de riñón derecho.
Discusión
En nuestro país, el cáncer renal representa el 1%-5% de todas las neoplasias4. El carcinoma de células claras representa más del 80% de los casos1, seguido por el carcinoma papilar y el carcinoma de células cromófobas2-4. Este último representa el 3%-5% de las neoplasias derivadas del epitelio tubular renal2,4,5,11, parece derivar de la porción cortical del túbulo colector4, se diagnostica principalmente en la 6° década de la vida5,11 y tiene una incidencia similar en hombres y mujeres5. El carcinoma de células cromófobas comprende un grupo heterogéneo que incluye los tipos clásico, eosinofílico y mixto3,5. El principal criterio diagnóstico es su morfología asociada a un inmunofenotipo característico (CK7 difuso y KIT positivo)5. El análisis genético muestra una deleción de los cromosomas 1, 2, 6, 10, 13, 17 y 212,4,5,11. El diagnóstico diferencial entre carcinoma de células cromófobas, carcinoma de células claras y oncocitoma representa un reto para el anatomopatólogo, ya que comparten características macroscópicas y microscópicas muy difíciles de diferenciar6,12,13, por lo tanto, el uso de técnicas diagnósticas auxiliares como la inmunohistoquímica, es de mucha utilidad para ayudar a esclarecer el diagnóstico en casos difíciles6.
La mayoría de los carcinoma de células cromófobas son esporádicos, pero ocasionalmente se encuentra asociado con el síndrome de Birt-Hogg-Dubé5, en el cual los pacientes desarrollan fibrofoliculomas cutáneos, quistes pulmonares, neumotórax espontáneo y tumores renales4,5,7. Los cánceres renales hereditarios comprenden el 3%-5% de los tumores renales4. En 2001, se describió un síndrome de cáncer renal familiar en el cual los pacientes comúnmente desarrollan leiomiomas cutáneos y uterinos, y carcinoma de células renales papilar de tipo 24,7-9. El gen afectado en el síndrome de leiomiomatosis uterina y cáncer renal hereditario es el gen de la fumarato hidratasa en el locus 1q42-44, con herencia autosómica dominante4,7. La penetrancia para el carcinoma de células renales es menor que para las manifestaciones cutáneas y uterinas14. Los leiomiomas cutáneos aparecen en el 76% de los individuos afectados15, los leiomiomas uterinos usualmente son múltiples, grandes15 y de inicio temprano; los tumores renales suelen ser unifocales, unilaterales8,15, aparecen en edad temprana7, son más agresivos que en otros síndromes de cáncer renal hereditario y van desde carcinoma de células renales papilar de tipo 2 a carcinomas del conducto colector15.
En la actualidad más del 50% de los carcinomas de células renales se diagnostica de manera incidental4,15. Los síntomas asociados pueden deberse al crecimiento tumoral local, a la enfermedad metastásica o a los síndromes paraneoplásicos, los cuales se observan en el 20% de los pacientes y se deben a la secreción anómala de las sustancias que normalmente produce el riñón4. Uno de los síndromes paraneoplásicos vinculado con el carcinoma de células renales es la disfunción hepática no metastásica o síndrome de Stauffer, que se ha comunicado en el 3%-20% de los casos. La función hepática se normaliza después de la nefrectomía en el 60%-70% de los casos4.
El carcinoma de células renales sigue siendo una enfermedad primordialmente quirúrgica, ya que todavía se le considera el paradigma del tumor quimiorrefractario. El objetivo de la terapia quirúrgica es escindir todo el tumor con un margen quirúrgico suficiente4. La cirugía de preservación de la nefrona consiste en la resección local completa de un tumor renal, al tiempo que se deja la mayor cantidad posible de parénquima funcional normal en el riñón afectado4,16. Esta modalidad quirúrgica tiene indicación en las situaciones en las cuales la nefrectomía radical dejaría al paciente anéfrico o con alto riesgo de la necesidad final de diálisis4, tal es el caso de los pacientes con masas renales en un riñón solitario16,17, aunque desde hace aproximadamente 15 años, en algunos centros, las indicaciones se han extendido a pacientes con tumores unilaterales pequeños (menores de 7 cm) y riñón contralateral normal15-17, esto debido a un mejor entendimiento de la histología del tumor renal y su amenaza oncológica, la equivalencia oncológica de la nefrectomía radical y la nefrectomía parcial en tumores renales T1 y la opinión emergente sobre la enfermedad renal crónica y su potencial efecto adverso en la salud cardiovascular15,18, ya que se ha propuesto que el estado de riñón solitario es el factor de riesgo más importante para desarrollar nefropatía crónica en estos pacientes19,20. Se sabe que el riesgo de desarrollar enfermedad renal crónica (definida como una TFG < 60 mL/min/1.73 m2)18 después de nefrectomía parcial, está en relación con el tiempo que permanece pinzada la arteria renal y con la cantidad de tejido resecado20,21. Un tiempo de isquemia caliente mayor de 20 minutos o de isquemia fría mayor de 35 minutos, se relaciona con una incidencia más alta de falla renal22,23. Los pacientes con una resección mayor del 50% de la masa renal pueden permanecer con función renal estable, pero se encuentran en mayor riesgo de desarrollar proteinuria, glomerulopatía y falla renal progresiva19. La modalidad quirúrgica depende de la capacidad del cirujano y puede realizarse de forma abierta, laparoscópica o laparoscópica asistida por robot17, aunque la modalidad laparoscópica se ha asociado con un mayor tiempo de isquemia y mayor prevalencia de daño renal crónico con la necesidad ulterior de diálisis15. Además, otras complicaciones asociadas a la nefrectomía parcial incluyen fístula urinaria, urinoma, absceso perinéfrico, trombosis arterial, trombosis venosa, estancia intrahospitalaria prolongada y sangrado tardío15,16,18-21.
La principal desventaja de la cirugía de preservación de la nefrona es el riesgo de recidiva local en el riñón operado4, la cual se presenta en el 10% de los pacientes con riñón solitario, contra 1%-6% en los pacientes con riñón contralateral normal19. En pacientes con tumores en un riñón solitario tratados con enucleación o nefrectomía parcial, con seguimiento a 5 y 10 años, la supervivencia global ha sido de 74% y 45%, la supervivencia específica de cáncer de 80% y 63%, la supervivencia libre de recurrencia local de 89% y 80%, y la supervivencia libre de metástasis de 69% y 50%, respectivamente19.
El tipo histológico del carcinoma renal tiene cierta influencia pronóstica. En términos generales, se considera que el carcinoma de células cromófobas tiene mejor pronóstico que el carcinoma de células claras1,2,5, con una supervivencia libre de enfermedad a los 5 años de 83.9% y a los 10 años de 77.9%5 y una mortalidad de 10%11. Sin embargo, el estadio tumoral, el tamaño de la neoplasia y el grado nuclear, siguen siendo los factores de mayor valor pronóstico2,3,5.
Existen datos contradictorios acerca de los efectos a largo plazo que trae consigo la disminución de la masa renal en los pacientes sometidos a nefrectomía unilateral21,24-26. no hay datos convincentes que indiquen que los donantes vivos corran un mayor riesgo a largo plazo debido a la donación renal. no obstante, se recomienda una evaluación de seguimiento periódica a largo plazo de los donantes. De ello puede encargarse el médico personal del donante10.
Conclusiones
Consideramos que es conveniente un seguimiento periódico a largo plazo de los pacientes con riñón único, ya que esto permite detectar de forma temprana cualquier patología que comprometa la función renal, pudiendo brindar tratamientos encaminados a conservar la mayor cantidad de nefronas.
Durante el estudio de la paciente no se encontraron datos clínicos o paraclínicos suficientes para integrar un síndrome de cáncer renal hereditario y leiomiomatosis uterina, en el cual el subtipo histopatológico suele ser papilar tipo 2 y no cromófobo, por lo tanto, se trata de una paciente con tumor renal cromófobo esporádico en riñón único asociado de manera incidental a miomatosis uterina. El manejo establecido no fue diferente de lo reportado en la literatura universal.
Los hallazgos de inmunohistoquímica en este caso están en consonancia con lo reportado en la literatura mundial. Los patrones encontrados en las tinciones de hematoxilina & eosina son muy orientadores para el diagnóstico de tumores cromófobos, sin embargo, estos hallazgos se ven reforzados por los resultados de los estudios de inmunohistoquímica.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Financiamiento
No se recibió patrocinio para llevar a cabo este artículo.
* Autor de correspondencia:
Departamento de Urología,
Hospital General del Estado.
Luis Encinas s/n, Colonia Centro,
C.P. 83000, Hermosillo, Son., México.
Teléfono: (662) 259 2500, ext. 2584.
Correo electrónico: elconejoblas2@hotmail.com (A. Blas-Reina).