


Relatar o caso de um recém‐nascido prematuro com íleo meconial complexo e fibrose cística.
Descrição do casoRecém‐nascido do sexo masculino nasceu de parto vaginal com 33 semanas e cinco dias de idade gestacional e apresentou desconforto respiratório e distensão abdominal grave. Foi submetido à laparotomia exploratória no primeiro dia de vida e identificado íleo meconial com peritonite secundária. Foram feitas ressecção ileal e ileostomia, com reconstrução do trânsito intestinal aos 20 dias de vida. Com 11 dias de idade, a primeira dosagem sérica de tripsina imunorreativa (TIR) foi 154ng/mL (valor de referência = 70) e optou‐se pelo início da terapia de reposição oral de enzimas pancreáticas. Após 23 dias, a segunda TIR foi 172ng/mL (valor de referência = 70). Recebeu alta com 35 dias de vida com encaminhamentos à rede básica de saúde e ao serviço de referência para a detecção de fibrose cística. Foi atendido no ambulatório de triagem neonatal para fibrose cística aos 65 dias de vida e apresentava desnutrição e desconforto respiratório. O resultado do teste do cloro no suor foi positivo (126 mEq/L).
ComentáriosO caso ilustra a rápida evolução da fibrose cística em um paciente prematuro com íleo meconial complexo como primeira manifestação clínica.
To report a case of a preterm infant with complex meconium ileus at birth and cystic fibrosis.
Case descriptionA male infant was born by vaginal delivery at 33 weeks and 5 days of gestational age with respiratory distress and severe abdominal distension. The exploratory laparotomy in the first day of life identified meconium ileus and secondary peritonitis. Ileal resection and ileostomy were performed, followed by reconstruction of the bowel transit at 20 days of life. At 11 days of life, the first immunoreactive trypsinogen (IRT) was 154ng/mL (reference value=70), and oral pancreatic enzymes replacement therapy was started. After 23 days, the second IRT was 172ng/mL (reference value=70). At 35 days of age he was discharged with referrals to primary care and to a special clinic for CF for the determination of sweat chloride. He was received in the outpatient clinic for neonatal screening for CF at 65 days of life presenting malnutrition and respiratory distress. The sweat chloride test was performed, with a positive result (126mEq/L).
CommentsThis case illustrates the rapid evolution of CF in a premature patient with complex Meconium ileus as the first clinical manifestation.
A fibrose cística (FC) é a doença recessiva autossômica letal mais prevalente e atinge 1:2.000 brancos. É causada pela alteração de um gene localizado no braço longo do cromossomo 7, que codifica uma proteína de 1.480 aminoácidos, reguladora da condutância transmembrana da fibrose cística (CFTR), que funciona como um canal de cloro na membrana apical das células epiteliais.1 Essa alteração resulta em uma mudança na viscosidade das secreções, com a produção de muco espesso, que leva principalmente à má absorção, perda de eletrólitos no suor e alteração das secreções pulmonares. Há mais de 1.900 mutações genéticas conhecidas, bem como genes modificadores da doença.2 Essa heterogeneidade fenotípica envolve diferentes apresentações clínicas, que variam de leve a grave, as quais podem determinar um resultado letal. A apresentação clássica da FC é a doença pulmonar crônica (infecções pulmonares recorrentes), insuficiência pancreática exócrina (diarreia e desnutrição), perda de sal e síndrome de azoospermia obstrutiva.3 Nos casos sem manifestações clínicas sugestivas de FC no primeiro mês de vida, a triagem neonatal pode levar à detecção precoce e o tratamento imediato da insuficiência pancreática, das deficiências nutricionais e do comprometimento pulmonar, melhora a sobrevida e facilita o desenvolvimento de estratégias de tratamento.4
Menos frequentemente, o íleo meconial (IM) pode ser a primeira manifestação da FC no período neonatal e ocorre em cerca de 20% dos pacientes com insuficiência pancreática. Esse quadro clínico é causado por obstrução do íleo terminal com mecônio espesso que contém grandes quantidades de proteína. O IM complexo é uma condição grave, significativamente mais frequente em pacientes sem FC de menor idade gestacional e peso ao nascer do que em pacientes com FC. O IM é classificado como complexo quando associado à perfuração ileal.5,6 Cerca de 80% dos casos de IM são devidos à FC e seria ideal fazer um teste precoce de cloro no suor antes de 48 horas de vida, embora isso não seja sempre viável.7,8 Crianças com IM parecem ter função pulmonar normal no diagnóstico da FC, com progressão mais lenta da doença pulmonar do que aqueles diagnosticados devido a sintomas respiratórios.9,10 No entanto, acredita‐se atualmente que a inflamação pulmonar pode ocorrer precocemente e até mesmo preceder o início da infecção em crianças com fibrose cística recém‐diagnosticada.11
O objetivo do presente relato é narrar o caso de uma criança com IM complexo que apresentou má evolução inicial, apesar da suspeita clínica de FC.
Descrição do casoUma criança do sexo masculino foi recebida no ambulatório de triagem neonatal para FC aos 65 dias de vida. O paciente nasceu por parto vaginal, pesava 2.100g com 33 semanas e cinco dias de idade gestacional, com escore de Apgar de 6 e 9 ao primeiro e quinto minuto. A mãe era uma primigesta de 19 anos, que havia feito nove consultas de pré‐natal, com sorologia negativa para infecções verticais e ultrassonografia obstétrica normal. Imediatamente após o nascimento, a criança foi levada para a unidade de cuidado intensivo neonatal devido a desconforto respiratório precoce e distensão abdominal grave. Uma radiografia simples de abdômen revelou ausência de ar na parte inferior. O paciente foi submetido a uma laparotomia exploradora, no primeiro dia de vida, que identificou IM e peritonite secundária à perfuração intestinal. Ressecção ileal e ileostomia foram feitas, seguidas por reconstrução do trânsito intestinal aos 20 dias de vida. Ele teve sepse neonatal precoce e tardia durante o período de internação e necessitou de antibioticoterapia prolongada. A nutrição parenteral foi gradualmente substituída por fórmula infantil até um volume de 160mL/kg/dia. Aos 11 dias de vida, o resultado da primeira tripsina imunorreativa (TIR) foi 154ng/mL e a terapia de substituição de enzimas pancreáticas foi iniciada (1.000UI/kg/dose por via oral a cada três horas antes da alimentação). Após 23 dias, a segunda TIR foi de 172ng/mL. Aos 35 dias de idade, ele pesava 2.040g e recebeu alta com encaminhamentos para os cuidados primários e para uma clínica especializada em FC, para a determinação do teste de cloro no suor.
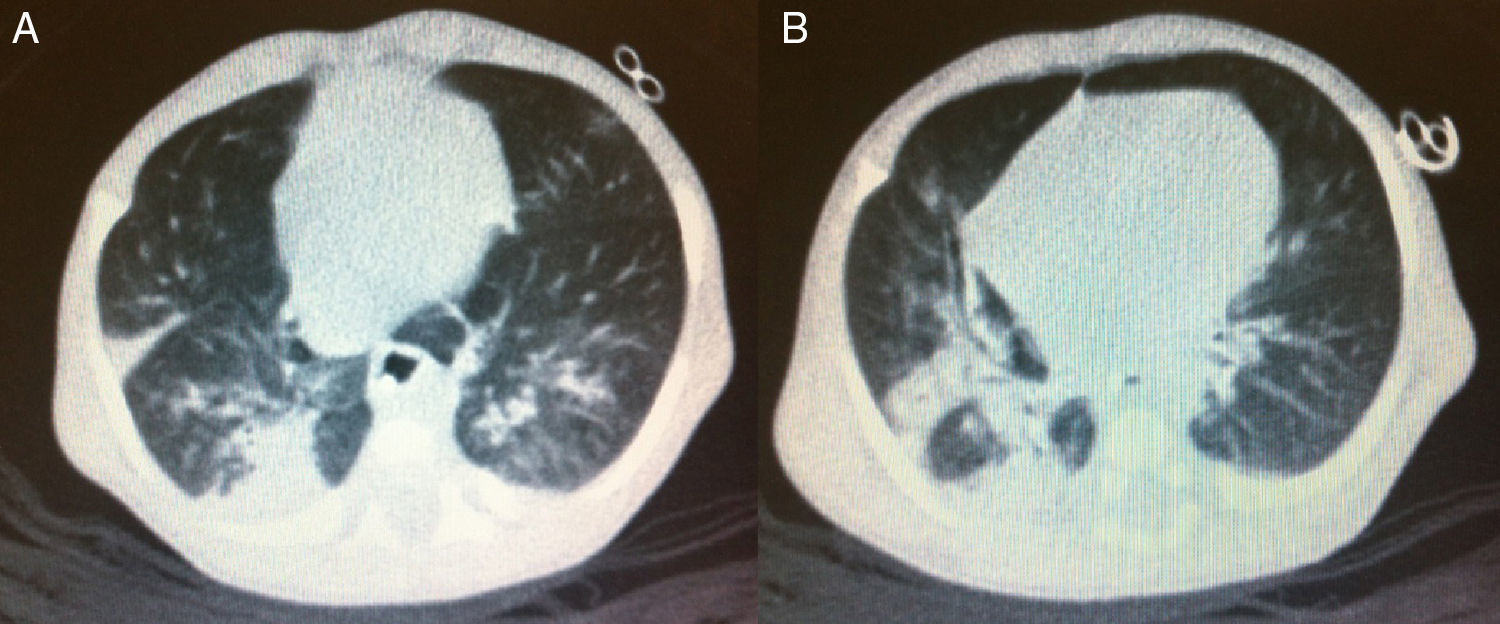
Aos 65 dias de vida, o teste de cloro no suor foi feito no serviço de referência com um resultado positivo (126 mEq/L). Nesse mesmo dia, o paciente foi atendido no ambulatório de seguimento para triagem neonatal de FC. De acordo com a mãe, as mesmas orientações recebidas à alta hospitalar ainda eram mantidas em relação ao volume de fórmula infantil e da dosagem de enzimas pancreáticas, as quais ela havia sido instruída a oferecer dissolvidas em água. Ele pesava 2.610g e seu comprimento era de 48 centímetros. Ele ganhou 19g/dia após a alta. Ele apresentava taquipneia (62 ciclos respiratórios/minuto), palidez (2/4+) e tosse seca persistente. Sua radiografia de tórax revelou hiperinflação pulmonar e condensação compatível com doença pneumônica. O paciente foi hospitalizado e os exames laboratoriais feitos durante a internação são apresentados na tabela 1. O teste de proteína C‐reativa (PCR) feito na admissão foi 18,8 mg/dL (valor de referência <0,5mg/dL por turbidimetria). Os testes de laboratório para vírus sincicial respiratório, adenovírus, influenza, assim como culturas de sangue, foram negativos. Uma tomografia computadorizada pulmonar foi feita no sexto dia de internação hospitalar (fig. 1).
 Mostra várias bandas de atelectasia com bronquiectasias e alguns brônquios com paredes espessas. B) Mostra outro espessamento da parede brônquica e inflamação peribronquial e extensa área de condensação subsequente.")
Uma dieta entérica foi prescrita com um volume de 150mL/kg/dia de fórmula semielementar à diluição de 1:25. A dose de enzimas pancreáticas foi ajustada para 5.000U antes da alimentação, a cada três horas. Uma transfusão de concentrado de hemácias foi feita. Sódio (4 mEq/kg/dia) e potássio (1mEq/kg/dia) foram administrados via enteral. Foram prescritas multivitaminas (24 gotas/dia) e sulfato de zinco (1mg/kg/dia). Após a cultura, e com base no microrganismo isolado da secreção do trato respiratório superior (tabela 1), foram prescritas ceftazidima (200mg/kg/dia), gentamicina (5mg/kg/dia), e oxacilina (200mg/kg/dia). Devido à ressecção do íleo, vitamina B12 (100mcg) foi administrada por via intramuscular. No sétimo dia de internação, devido à pioria progressiva do padrão respiratório, ventilação não invasiva (CPAP) foi iniciada e mantida por duas semanas. Após a estabilização da doença pulmonar, o paciente evoluiu com ganho de peso satisfatório. O paciente recebeu alta após 40 dias, com 3.490g, sem antibióticos, com reposição de sódio (ajustado para 2 mEq/kg/dia) e sulfato ferroso (4mg/kg/dia).
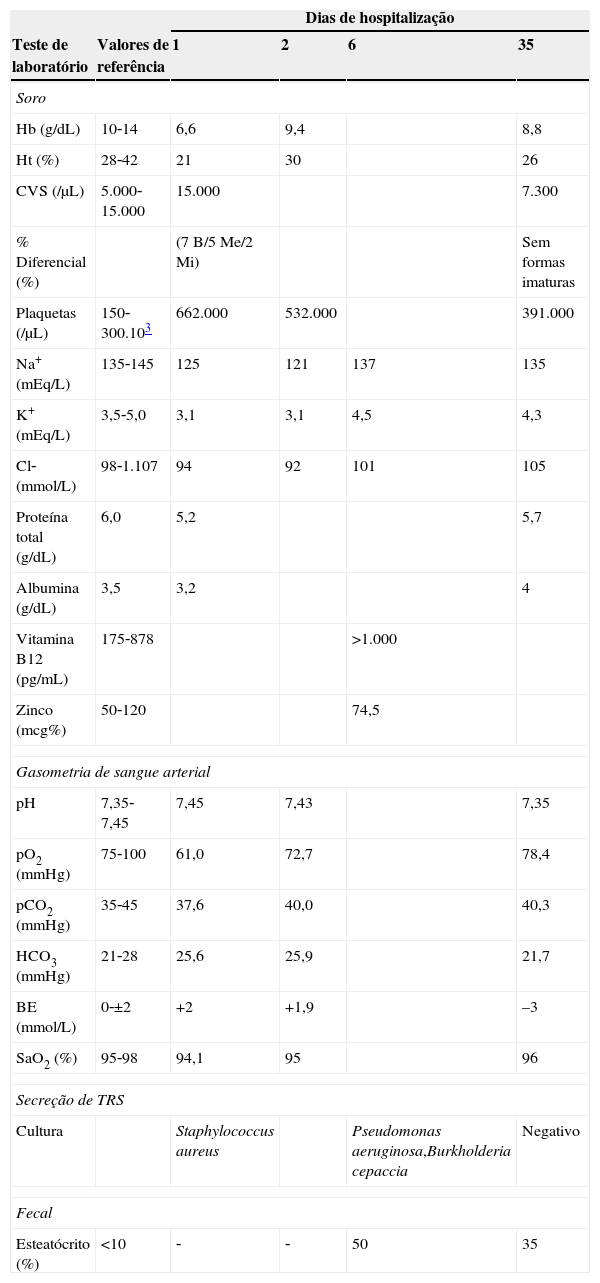
Exames feitos no início e no fim da internação do paciente no serviço de referência para tratamento de FC
| Dias de hospitalização | |||||
|---|---|---|---|---|---|
| Teste de laboratório | Valores de referência | 1 | 2 | 6 | 35 |
| Soro | |||||
| Hb (g/dL) | 10‐14 | 6,6 | 9,4 | 8,8 | |
| Ht (%) | 28‐42 | 21 | 30 | 26 | |
| CVS (/μL) | 5.000‐15.000 | 15.000 | 7.300 | ||
| % Diferencial (%) | (7 B/5 Me/2 Mi) | Sem formas imaturas | |||
| Plaquetas (/μL) | 150‐300.103 | 662.000 | 532.000 | 391.000 | |
| Na+ (mEq/L) | 135‐145 | 125 | 121 | 137 | 135 |
| K+ (mEq/L) | 3,5‐5,0 | 3,1 | 3,1 | 4,5 | 4,3 |
| Cl‐ (mmol/L) | 98‐1.107 | 94 | 92 | 101 | 105 |
| Proteína total (g/dL) | 6,0 | 5,2 | 5,7 | ||
| Albumina (g/dL) | 3,5 | 3,2 | 4 | ||
| Vitamina B12 (pg/mL) | 175‐878 | >1.000 | |||
| Zinco (mcg%) | 50‐120 | 74,5 | |||
| Gasometria de sangue arterial | |||||
| pH | 7,35‐7,45 | 7,45 | 7,43 | 7,35 | |
| pO2 (mmHg) | 75‐100 | 61,0 | 72,7 | 78,4 | |
| pCO2 (mmHg) | 35‐45 | 37,6 | 40,0 | 40,3 | |
| HCO3 (mmHg) | 21‐28 | 25,6 | 25,9 | 21,7 | |
| BE (mmol/L) | 0‐±2 | +2 | +1,9 | –3 | |
| SaO2 (%) | 95‐98 | 94,1 | 95 | 96 | |
| Secreção de TRS | |||||
| Cultura | Staphylococcus aureus | Pseudomonas aeruginosa,Burkholderia cepaccia | Negativo | ||
| Fecal | |||||
| Esteatócrito (%) | <10 | ‐ | ‐ | 50 | 35 |
B, bands; Me, metamielócitos; Mi, mielócitos; TRS, trato respiratório superior.
Sabe‐se que a confirmação de FC pode ser difícil durante os primeiros dias de vida, mas na presença de íleo meconial esse diagnóstico diferencial deve obrigatoriamente ser considerado. O teste de cloro no suor pode ser feito após 48 horas de vida, mas resultados mais confiáveis podem ser obtidos a partir da segunda semana de vida, com o paciente com peso superior a 2kg, com hidratação adequada e sem doença sistêmica significativa.12 O teste de cloro no suor é o padrão ouro para o diagnóstico da FC; no entanto, algumas situações podem alterar os resultados, tais como desidratação, baixo peso, erupção cutânea ou um estado geral ruim.13,14 A despeito da importância do teste de cloro no suor precoce, o estado clínico desse paciente não era apropriado para submetê‐lo ao teste durante os primeiros dias de vida.
A análise genética do regulador da condutância transmembrana na fibrose cística (CFTR) também ajuda o diagnóstico da FC quando detecta duas mutações conhecidas.15 O exame teria sido útil para esse paciente se tivesse detectado as duas mutações características para FC; no entanto, o exame não está rotineiramente disponível em nosso centro devido ao seu alto custo. O Programa Nacional de Triagem Neonatal em todo o país começou em 2011, apesar de ter sido implantado pela primeira vez em 2010, no Estado de São Paulo.16 O método de duas amostras de TIR foi adotado, com a primeira amostra colhida entre três e sete dias de vida e a segunda até 30 dias de vida. Se ambas as amostras (TIR) forem positivas (valor de referência ≥70ng/mL), a FC é confirmada por dois testes positivos de cloro no suor (valor de referência ≥60mEq/L). Um teste de cloro no suor entre 30 e 60mEq/L é limítrofe para recém‐nascidos e não exclui ou confirma imediatamente a doença.17
Apesar da importância de duas avaliações neonatais de TIRs para detecção da FC, o benefício parece menor para crianças com íleo meconial, porque recém‐nascidos com FC e IM podem apresentar valores iniciais de TIR baixos.18 Mesmo que os níveis de TIR permaneçam elevados, isso seria simplesmente um alerta para a possível presença de FC, sem confirmar a doença, pois vários fatores aumentam a possibilidade de valores falso‐positivos de TIR, incluindo o estresse perinatal.19 Embora fossem esperados valores baixos de TIR para o paciente em questão, os níveis detectados foram elevados, o que justificou a suspeita de FC e, provavelmente, também a decisão de iniciar empiricamente o tratamento de insuficiência pancreática devido à FC.
Recém‐nascidos com IM devem receber tratamento específico para a insuficiência pancreática enquanto a confirmação da FC não é feita pelo teste de cloro no suor. O ganho de peso durante a internação indica uma boa resposta.20 Relatos atuais afirmam que o IM não é considerado como fator de prognóstico ruim em pacientes com tratamento eficaz para FC.21 O presente paciente teve alta recebendo enzimas pancreáticas, com a mãe dissolvendo‐as em água. Esse procedimento ilustra a importância de um acompanhamento precoce e frequente de crianças com suspeita de uma doença crônica, rara e grave pelo serviço de referência para tratamento de FC, tanto em relação ao apoio à família quanto ao aprimoramento das orientações oferecidas, de modo que sejam rigorosamente seguidas. Deve‐se ressaltar que esse tipo de cuidado envolve, mas não substitui o monitoramento do paciente por um pediatra clínico geral. O tratamento da insuficiência pancreática envolve a ingestão oral de microesferas intactas de enzimas pancreáticas imediatamente antes da alimentação, com variação de 2.000 a 4.000UI de lipase para cada 120mL de fórmula ou leite materno. Embora menos fisiológico, o cálculo também pode ser feito com uma dose de 1.000UI de lipase/kg/refeição para crianças com idade inferior a quatro anos. Evitam‐se doses mais elevadas do que 2.500 UI/kg/refeição e 10.000UI/kg/dia, o que poderia provocar colonopatia fibrosante.22
Os pulmões podem ser afetados desde o período da triagem neonatal para FC; 81% dos casos mostram anormalidades estruturais, 45% espessamento das paredes brônquicas e 21% infecção pulmonar.23 Tosse e dispneia em neonatos e lactentes indicam a necessidade de incluir a FC na lista de diagnósticos diferenciais. Inflamação ou infecção pulmonar já pode estar presente. O Staphylococcus aureus é o microrganismo mais frequentemente detectado, seguido por Pseudomonas aeruginosa, com sintomas respiratórios significativos. Os investigadores observaram que os lactentes com fibrose cística detectada por meio da triagem neonatal podem ter doença pulmonar associada à infecção bacteriana desde os primeiros dias de vida.24,25 Essa é uma fonte de preocupação, uma vez que está ligada ao aparecimento precoce de bronquiectasia e inflamação pulmonar mais grave.
O presente relato de caso ilustra a evolução rápida do múltiplo sistema da FC em um paciente prematuro com IM complexo como primeira manifestação clínica. Ele também mostra a presença de resultados paradoxais de triagem neonatal (TIR/TIR) em pacientes com IM e as dificuldades do teste precoce de cloro no suor e de genotipagem para FC em casos de IM. Além disso, ressalta‐se a importância de um rápido encaminhamento de pacientes com IM e suspeita de FC para uma equipe de especialistas.
FinanciamentoO estudo não recebeu financiamento.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
