


Pseudohypoparathyroidism is a genetically heterogeneous condition characterized by hypocalcemia and hyperphosphatemia resulting from end-organ resistance to parathyroid hormone (PTH). It is classified into several distinct entities (type Ia, Ib, Ic, type II), according to the molecular causes and clinical features of the patients.
Case reportWe report a symptomatic case of a pediatric patient with hypocalcaemia, hyperphosphatemia, hypomagnesaemia and an elevated serum PTH level that presented with tetany. The molecular genetic test revealed a GNAS gene methylation defect of the A/B promoter, which confirms the diagnosis of a sporadic form of pseudohypoparathyroidism type Ib.
o pseudohipoparatiroidismo é uma doença geneticamente heterogénea que se caracteriza por hipocalcemia e hiperfosfatemia que têm origem numa resistência periférica dos órgãos-alvo a hormona paratiroideia (PTH). Classifica-se em várias entidades distintas (tipo Ia, Ib, Ic e tipo II) de acordo com a etiologia molecular e características clínicas dos doentes.
Caso clínicoos autores descrevem o caso de uma criança com hipocalcemia, hiperfosfatemia, hipomagnesemia e elevação da PTH sérica que se manifestou através de tetania. O estudo molecular revelou um defeito de metilação no promoter A/B do gene GNAS que confirmou o diagnóstico de uma forma esporádica de pseudohipoparatiroidismo tipo Ib.
Pseudohypoparathyroidism (PHP) is a genetically heterogeneous condition characterized by hypocalcemia and hyperphosphatemia resulting from end-organ resistance to parathyroid hormone (PTH).1 Patients with this condition carry mutations within the GNAS locus on chromosome 20q13.3.2,3 PHP is classified into several distinct entities (type Ia, Ib, Ic, type II), according to the molecular causes and clinical features of the patients.4
The GNAS gene is paternally imprinted in the proximal renal tubules in humans. PHP-Ib is caused by defects of the maternally derived GNAS allele and patients show hypomethylation at one or more of the 4 differentially methylated regions (DMRs) of GNAS. PHP-Ib can follow an autosomal dominant trait (AD-PHP-Ib) or occur as a disorder that appears to be sporadic.5–7 Genetic causes of PHP-Ib include cryptic deletions within the genes neighboring GNAS, STX16 and NESP55, and epimutation of GNAS DMRs.5,8 A few of the sporadic patients, all with broad methylation changes, were shown to be affected by paternal uniparental isodisomy (patUPD20) involving the long arm of chromosome 20, which comprises the GNAS locus,9–11 but most apparently sporadic cases have not yet been resolved at the molecular level5,8,12–16 raising the question as to whether alternative mechanisms could explain the methylation changes observed in sporadic PHP-Ib patients such as environmental and exogenous factors.17–20
PHP-Ib patients present with PTH- and, sometimes, TSH-resistance and typically lack the features of Albright hereditary osteodystrophy (AHO), although some PHP-Ib cases with GNAS imprinting defects have recently been shown to have AHO features.21–24 These patients are usually identified by hypocalcemia-associated neuromuscular irritability, such as tetany, generalized convulsions, and/or muscle cramps, although a substantial fraction of the patients remain asymptomatic and are identified only by familial studies.6,25
Here, we report a Portuguese patient with sporadic PHP-Ib due to promoter A/B methylation defect with homozygous abnormality.
Case reportThis female patient was born as the first child to nonconsanguineous parents from Guinea-Bissau at 41 weeks of gestation after an uncomplicated pregnancy and delivery. Her birth weight was 4550g (95th percentile), length 55cm (95th percentile) and head circumference 36.5cm (83rd percentile). She developed neonatal hypoglycemia immediately after birth and hyperbilirrubinemia on the 3rd day of life. Neonatal screening tests were normal and her postnatal growth evolved on the 95th percentile with no developmental delay.
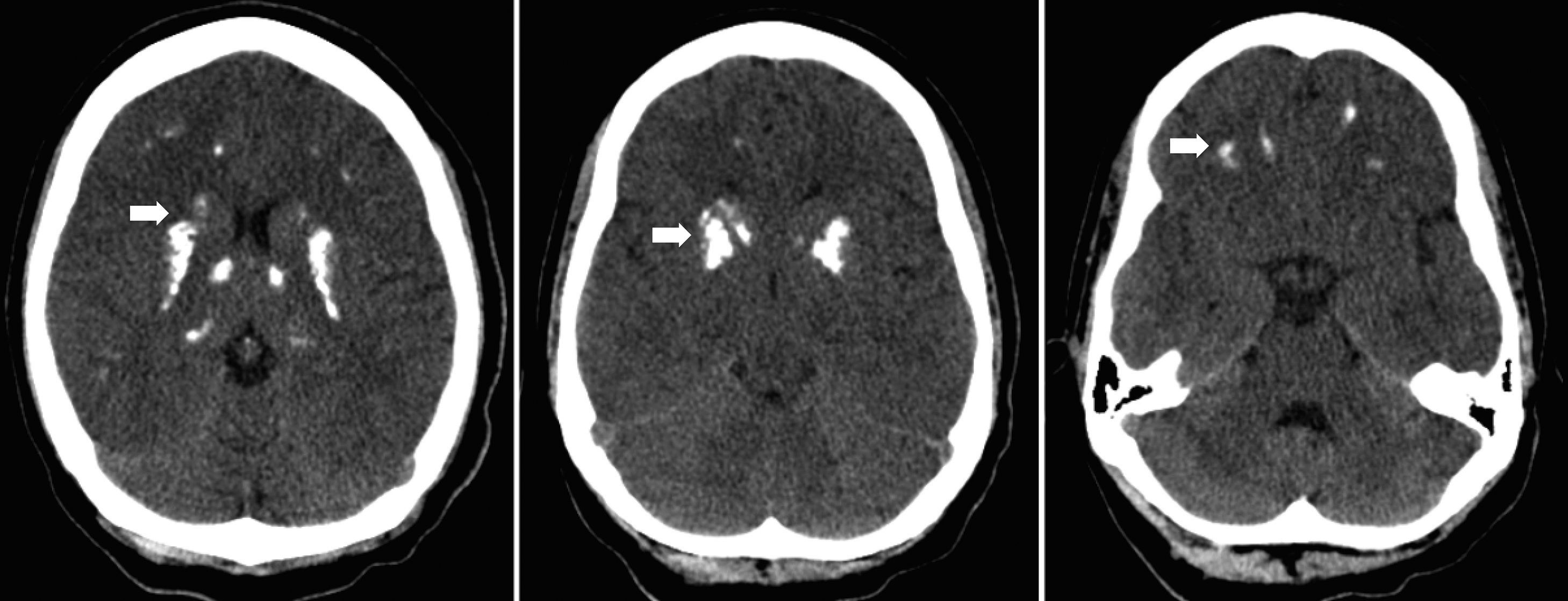
In December 2002, at the age of nine she was admitted to the hospital due to tetany, which was wrongly interpreted as a generalized seizure. There was a history of general fatigue, poor school performance and tingling in the hands that began 2 months earlier. Physical examination showed truncal obesity, round face but no other phenotypic features of AHO. Her height was 143cm (93rd percentile). Laboratory investigation revealed serum hypocalcemia with a calcium level of 5.6mg/dL (normal range 9.6–10.6mg/dL), hyperphosphatemia, hypomagnesaemia and an elevated serum PTH level of 165pg/mL (normal range 10–60pg/mL), normal alkaline phosphatase, serum albumin and a normal thyroid function. Head computerized tomography revealed bilateral, symmetric calcifications in thalamus, basal ganglia and white subcortical frontotemporal substance (Fig. 1).
.")
She was treated with oral calcium and calcitriol and her general fatigue markedly improved.
Her mother and younger sister did not show clinical or laboratory evidence suggestive of an abnormal regulation of calcium and phosphate homeostasis.
She was referred to a geneticist for further evaluation. After obtaining written informed consent, genomic DNA was extracted from leukocytes of our patient by standard methods. Methylation analysis of the promoter regions of the splice variants XLαs and A/B was performed by bisulfite sequencing.26 The molecular genetic result revealed a GNAS gene methylation defect of the A/B promoter, which is specific for PHP type Ib. The STX16 deletion was not detectable in the patient.
At her follow-up, at the age of 17 the patient remained without symptoms, having normal puberty, height 173cm (94th percentile) and obesity with a BMI of 31.24kg/m2. Currently her regimen consists of both calcitriol and calcium supplements with poor adherence to treatment. Her serum calcium levels have ranged from 6.4 to 8.0mg/dL (normal range 9.6–10.6mg/dL) with phosphorous levels varying from 6.8 to 7.3mg/dL (normal range 3.1–4.7mg/dL) and PTH levels above the normal range. Her urinary calcium to creatinine ratios have been normal. She has had no further episodes of tetany and has shown both clinical and biochemical improvement.
At the age of 18 she became pregnant. At 32 weeks of pregnancy she was hospitalized for vaginal bleed, was treated with oral iron and rest was advised. The delivery, at 38 weeks, was uneventful. Her asymptomatic son was also referred to a geneticist and the molecular genetic test performed at the age of 7 months did not confirm the diagnosis of PHP type Ib.
DiscussionIn this report, we describe laboratory, epigenetic, and genetic findings in a patient, who presented with tetany, hypocalcemia, hyperphosphatemia, hypomagnesaemia and an elevated serum PTH level at the age of nine. Although the initial clinical onset of PHP-Ib may be due to a severe hypocalcaemia leading to neuromuscular irritability, such as tetany or generalized convulsions, the hypocalcaemic condition may become clinically evident only during the pubertal growth spurt, when calcium requirements are higher. Another important factor to be considered is the seasonal period. It is usually during autumn or winter, when the sun exposure is lower, that the 25-hydroxyvitamin D reaches the lowest blood concentration that might lead to hypocalcaemia, which is consistent with the time of presentation of our patient. Another possible explanation for the late detection of hypocalcaemia might be a delayed onset of parathyroid hormone-resistance due a gradual development of paternal Gs-alpha silencing in target tissues.27
The combination of high PTH and low serum calcium led to the suspicion of end organ resistance to PTH. The mild features of AHO phenotype favored the diagnosis of PHP-Ia although her physical examination was not positive for short stature, heterotopic ossifications or mental retardation. Some PHP-Ib cases described have been shown to have some AHO features.21,23 Molecular characterization is currently a reliable method to differentiate the various subtypes of PHP. The diagnosis of PHP-Ib in our patient was confirmed by methylation analysis of the promoter regions of the splice variants XLαs and A/B, which revealed a GNAS gene methylation defect. The STX16 deletion was not detectable in the patient and a sporadic form of PHP-Ib was confirmed.
Her healthy mother and younger sister were not tested for the genetic defect. PHP-Ib is transmitted only from the mother and her son's molecular genetic test was negative for the condition. The decision to request the genetic test despite our patient's sporadic form was based on the fact that the identification of this rare disease allows an early diagnosis, and may prevent hypocalcemia-related life-threatening complications.
The aim of PHP therapy is to obtain an adequate calcium-phosphate control and to correct the multiple hormonal resistances, when present. The goal is to maintain blood calcium between 8.8 and 10.8mg/dL, and the urinary calcium/urinary creatinine ratio <0.228 which was not the case in our patient due to poor treatment adherence. It is crucial to emphasize the importance of treatment adherence that may prevent further symptoms.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank Dr. Mato Nagel (Center for Nephrology and Metabolic Disorders, Germany) for the technical assistance.
