


A acromegalia é uma doença crónica rara, causada pela hipersecreção de hormona do crescimento (GH) e do fator de crescimento semelhante à insulina tipo 1 (IGF‐1). O diagnóstico da doença o mais precocemente possível é essencial para prevenir as suas complicações e morbilidade. Apresentamos o caso de uma mulher, enviada à nossa consulta de endocrinologia por obesidade, com escassas características acromegaloides, mas com várias comorbilidades desta patologia. Apesar do tempo de evolução até ao diagnóstico, do tamanho do tumor e de se tratar de um tumor misto, a doente está, neste momento, curada.
Acromegaly is a rare chronic disorder due to growth hormone (GH) hypersecretion and elevated levels of insulin growth factor‐1 (IGF‐1). An early diagnosis is crucial to prevent the complications and morbidity of the disease. We present a case of a female, sent to our outpatient department due to obesity, with few acromegalic features, but with several co‐morbidities of the disease. Despite of the delay in the diagnosis, the size of the tumor and the mixed cell immunocytochemistry, she was successfully treated.
A acromegalia é uma doença crónica rara, causada pelo excesso de produção de hormona do crescimento (GH), geralmente por um adenoma hipofisário. Muitas das manifestações clínicas da doença são comuns a outras patologias mais frequentes, o que faz com que o diagnóstico da acromegalia seja muitas vezes tardio, podendo a doença estar ativa sem ser reconhecida durante vários anos (10‐15 anos, segundo alguns autores)1. A acromegalia tem uma incidência, aproximada, de 5 casos/milhão/ano e uma prevalência de 60 casos por milhão de pessoas2. Em Portugal, a incidência da doença rondará os 50 novos casos por ano. Os adenomas da hipófise correspondem a 15% de todos os tumores intracranianos, sendo que 90% dos doentes com acromegalia terão um adenoma hipofisário produtor de GH, um macroadenoma em 70% dos casos3. Estes tumores podem produzir apenas GH isoladamente ou em associação a outras hormonas, sendo a mais frequente a prolactina (PRL). A doença acomete igualmente ambos os sexos, com maior incidência entre a 3.a e 4.a décadas de vida4.
As manifestações clínicas da acromegalia são muito variadas, dependem dos níveis de GH e fator de crescimento semelhante à insulina tipo 1 (IGF‐1), da idade do doente, do tamanho do tumor e do tempo decorrido entre o início da doença e o seu diagnóstico. Incluem sinais e sintomas associados ao efeito de massa do tumor hipofisário e efeitos sistémicos do excesso de GH e IGF‐1, nomeadamente doença cardiovascular, alterações metabólicas, manifestações respiratórias, ósseas e articulares, manifestações gastrointestinais, bem como outras manifestações endócrinas (bócio, hipercalciúria, diminuição da líbido, irregularidades menstruais)5.
As alterações somáticas e da aparência física são muito sugestivas da doença, contudo, podem ter um desenvolvimento muito indolente e insidioso, por vezes ao longo de anos, não sendo, nestes casos, imediatamente percetíveis. Em alguns doentes, estas alterações fenotípicas são tão subtis que não são percebidas pelo próprio nem pelos seus familiares, sendo também de difícil valorização para o clínico. Mesmo na ausência de características físicas sugestivas da doença, a acromegalia deve ser considerada um diagnóstico diferencial quando coexistem diversas patologias que podem ser explicadas por esta doença.
A acromegalia está associada a um aumento da mortalidade, de cerca de 2‐3 vezes, em relação à população geral, sendo esta, sobretudo, secundária a doença cardiovascular (60%), respiratória (25%) ou neoplásica (15%)6.
Caso clínicoMulher de 40 anos, natural de Santa Maria da Feira, referenciada à consulta de endocrinologia do nosso hospital por obesidade de grau III (IMC 40,4Kg/m2). Tinha ainda os diagnósticos de dislipidemia e diabetes mellitus tipo 2, estando medicada com sinvastatina e metformina. Nos antecedentes pessoais, destacava‐se uma amigdalectomia por hipertrofia amigdalina há 4 anos e síndrome do canal cárpico à direita, tendo sido submetida a cirurgia de descompressão. Quatro anos antes da primeira consulta no nosso hospital, no contexto de queixas de cefaleias intensas, foi efetuada uma tomografia axial computorizada cranioencefálica (TAC‐CE) que terá mostrado um macroadenoma hipofisário. Foi estudada na consulta de endocrinologia de outra unidade hospitalar, estudo esse a que não tivemos acesso, e teve alta com o diagnóstico de adenoma hipofisário não funcionante. A doente manteve queixas de cefaleias, entretanto mais intensas e frequentes, com várias vindas ao serviço de urgência hospitalar para analgesia endovenosa (EV), e artralgias. Referia ainda oligomenorreia, sendo que não estava a fazer qualquer terapêutica hormonal. Negava necessidade de aumentar o número dos sapatos, bem como necessidade de alargar os anéis que usava habitualmente. À observação, apresentava apenas discreto aumento do volume dos lábios e alargamento do nariz, embora a doente negasse alteração dos mesmos.
Apresentava‐se normotensa (122/70mmHg) e destacava‐se uma obesidade de grau III (IMC 40,4Kg/m2).
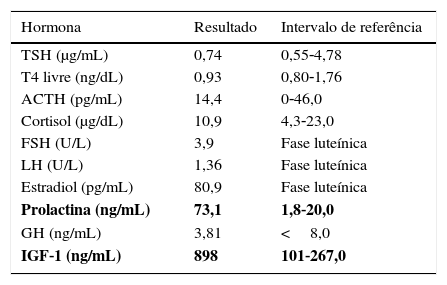
Apesar de a doente não apresentar alterações físicas sugestivas de acromegalia, perante a coexistência de diabetes mellitus, obesidade, dislipidemia, história de hipertofia amigdalina, síndrome do túnel cárpico, cefaleias e informação prévia de macroadenoma hipofisário, esta hipótese foi considerada. Foi pedida uma avaliação laboratorial completa do eixo hipofisário, que mostrou aumento de GH e IGF‐1 (3,81ng/ml e 898ng/mL respetivamente) e PRL também elevada (73,1ng/mL) (tabela 1).
Avaliação analítica hormonal efetuada na primeira consulta, mostrando aumento de IGF‐1 e PRL
| Hormona | Resultado | Intervalo de referência |
|---|---|---|
| TSH (μg/mL) | 0,74 | 0,55‐4,78 |
| T4 livre (ng/dL) | 0,93 | 0,80‐1,76 |
| ACTH (pg/mL) | 14,4 | 0‐46,0 |
| Cortisol (μg/dL) | 10,9 | 4,3‐23,0 |
| FSH (U/L) | 3,9 | Fase luteínica |
| LH (U/L) | 1,36 | Fase luteínica |
| Estradiol (pg/mL) | 80,9 | Fase luteínica |
| Prolactina (ng/mL) | 73,1 | 1,8‐20,0 |
| GH (ng/mL) | 3,81 | <8,0 |
| IGF‐1 (ng/mL) | 898 | 101‐267,0 |
Foi efetuada uma ressonância magnética cranioencefálica (RM‐CE), que mostrou um macroadenoma hipofisário (13,9mm×8,5mm×12,1mm) com necrose do núcleo central, sem evidência de invasão ou compressão das estruturas vizinhas. Procedeu‐se ao doseamento de GH após a prova de tolerância à glucose oral (PTGO), que confirmou o diagnóstico de acromegalia. O ecocardiograma transtorácico não mostrou alterações, nem a colonoscopia. Foi observada em consulta de neuro‐oftalmologia, não se tendo verificado alterações, nomeadamente no que concerne aos campos visuais.
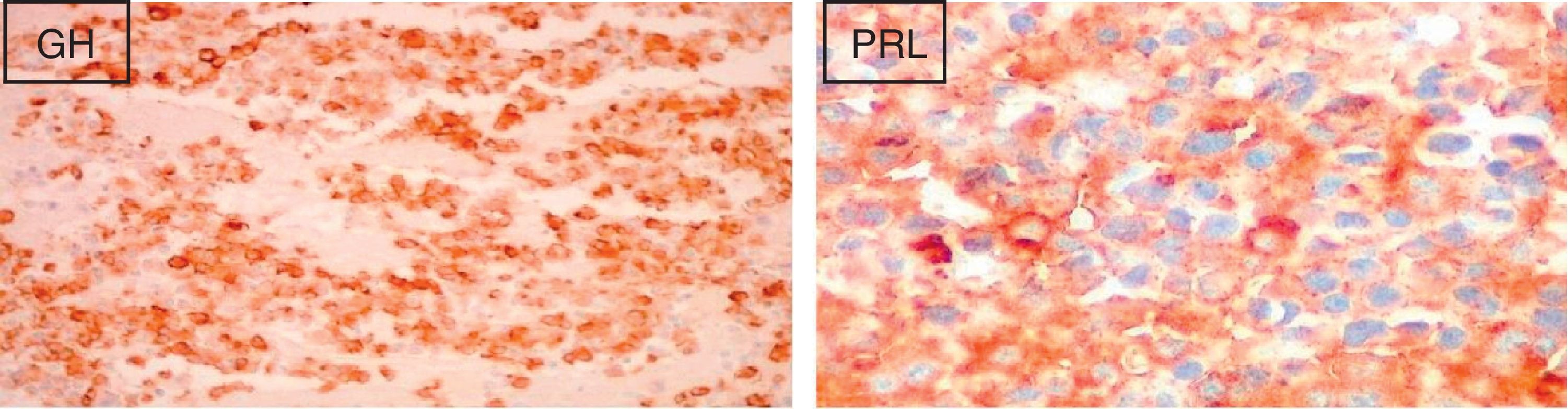
Foi iniciada terapêutica com octreotido 20mg, uma vez por mês, e a doente foi proposta para cirurgia. Ainda antes desta, e após 3 administrações de octreotido, verificou‐se uma importante diminuição do valor de IGF‐1 (378ng/ml) e normalização do valor de PRL. A doente foi submetida a excisão do adenoma hipofisário por via transesfenoidal, complicada pelo aparecimento de diabetes insípida transitória. O exame anatomopatológico da peça operatória mostrou um adenoma hipofisário sólido, com um índice proliferativo <3% e sem mitoses. A imuno‐histoquímica foi positiva para GH e PRL (fig. 1).

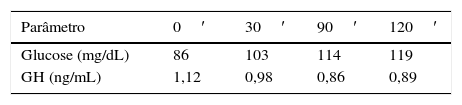
Doze semanas após a cirurgia, estava bioquimicamente controlada (IGF‐1 263ng/mL, GH 1,32), verificando‐se resolução total das cefaleias. Foi realizada RM‐CE de controlo, que não mostrou tecido tumoral, sendo que, 18 meses após a cirurgia, mantém níveis normais de IGF‐1 (270ng/mL) e PRL e PTGO com critérios de remissão cirúrgica da doença (GH<1,0ng/mL) (tabela 2). Além da remissão total das cefaleias e das artralgias, verificou‐se normalização do metabolismo da glucose e lipídico, bem como regularização dos ciclos menstruais.
DiscussãoO diagnóstico da acromegalia pode ser um desafio clínico. Embora óbvio quando são exuberantes as alterações fenotípicas características da doença, pode ser difícil e tardio quando tal não se verifica. Esta doente não tinha, de facto, a aparência característica da acromegalia, mas apresentava uma panóplia de manifestações da doença, nomeadamente cefaleias, diabetes, dislipidemia, obesidade, oligomenorreia e síndrome do canal cárpico. Acresce que muitas das queixas e patologias acima descritas são comuns e estão frequentemente associadas à obesidade, o que constitui um fator de confusão adicional. Além da história clínica detalhada, a informação do diagnóstico prévio de macroadenoma hipofisário, classificado como não funcionante, foi essencial para a suspeita diagnóstica, pois, de outra forma e na ausência do fenótipo característico de acromegalia, todas as queixas e diagnósticos da doente poderiam ser interpretados no contexto de uma obesidade de grau III.
As cefaleias eram a queixa principal e mais incapacitante da doente, sendo que cerca de 70% dos doentes com acromegalia referem queixas de cefaleias. A relação entre cefaleias e tumores hipofisários permanece discutível, sendo que ao longo do tempo têm sido propostos vários mecanismos para explicar esta associação (mecanismos mecânicos, bioquímicos, vasculares e biopsicossociais)7. Os mecanismos mecânicos relacionam‐se diretamente com a dimensão tumor e, embora esta doente tivesse um macroadenoma, não havia compressão ou invasão de estruturas vizinhas. Especula‐se que a própria GH, quando em excesso, possa induzir cefaleias. A suportar esta teoria está o facto de as cefaleias melhorarem significativamente após a cirurgia e serem um dos primeiros sintomas quando há recidiva da doença8. Paralelamente, alguns estudos mostram que crianças com défice de GH, sob terapêutica de substituição com esta hormona, referem frequentemente queixas de cefaleias coincidentes com o início da terapêutica9. Adicionalmente, os análogos da somatostatina têm demonstrado grande eficácia na melhoria deste sintoma10. No caso clínico em questão, a cefaleia foi o primeiro sintoma da doença, tendo melhorado enormemente após o início de terapêutica com análogo da somatostatina e resolvido completamente após a cirurgia, quer pela dimensão da massa tumoral quer pela diminuição da GH.
O tratamento de primeira linha na acromegalia é a cirurgia11. A terapêutica médica deve ser utilizada como adjuvante nos doentes com evidência de doença residual após a cirurgia e pode constituir a terapêutica inicial nos doentes que não são candidatos a cirurgia, quer por razões médicas quer por razões diretamente relacionadas com as características do tumor (adenomas com extensão extrasselar, nomeadamente invasão do seio cavernoso)11–13. Esta doente foi, logo após o diagnóstico e antes da cirurgia, medicada com octreotido. De facto, alguns estudos sugerem que a terapêutica com análogos da somatostatina antes da cirurgia diminui o risco cirúrgico e aumenta a probabilidade de controlo bioquímico da doença após a cirurgia14,15.
Tal como verificado neste caso clínico, muitos doentes com acromegalia têm adenomas produtores, não só de GH, mas também de PRL (15‐30%). Este facto não é negligenciável, já que, quando comparados com doentes com adenomas secretores apenas de GH, estes doentes têm a doença numa idade mais precoce, menos características acromegaloides, valores mais baixos de GH, mas tumores hipofisários de maiores dimensões. Alguns estudos sugerem ainda que doentes com adenomas secretores de GH e PRL têm uma menor probabilidade de cura e uma maior probabilidade de recidiva da doença, relativamente aos adenomas que produzem apenas GH16,17.
De forma a confirmar a remissão cirúrgica da doença, recomenda‐se a medição da IGF‐1 aproximadamente 12 semanas após a cirurgia, bem como nova RM‐CE e PTGO. Caso se confirme a remissão, o doseamento de IGF‐1 e a PTGO devem ser efetuados anualmente. Um valor de GH <2ng/mL no primeiro dia de pós‐operatório, assim como valores de GH <0,4ng/mL na PTGO, correlacionam‐se com uma maior probabilidade de remissão a longo prazo11, sendo, contudo, a remissão bioquímica da doença comprovada por valores de GH <1ng/mL na PTGO.
Apesar de este caso reportar um macroadenoma produtor de GH e PRL e, por isso, com maior probabilidade de persistência de doença ou recidiva da mesma, 18 meses após a cirurgia e sem qualquer terapêutica adjuvante, a doente apresenta um valor de IGF‐1 normal, bem como uma PTGO compatível com remissão cirúrgica da doença. A PRL é normal. Também as repercussões metabólicas da doença regrediram.
ConclusãoO diagnóstico de acromegalia nem sempre é óbvio e pode ser desafiante, sobretudo quando as alterações fenotípicas características da doença não são evidentes. Muitos dos sintomas e comorbilidades da acromegalia são comuns a outras patologias, nomeadamente à obesidade. Tal facto, como se verificou neste caso, pode atrasar o diagnóstico da doença, com as consequências que daí advêm. A colheita de uma história clínica completa e cuidada, e um elevado nível de suspeição são essenciais para o atempado e adequado diagnóstico da acromegalia e subsequente tratamento, com redução da morbilidade e mortalidade associadas à doença.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram ter recebido consentimento escrito dos pacientes e/ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Este caso clínico foi apresentado no ENEA 2014, em Sofia, com o apoio de uma bolsa atribuída pela SPEDM, que muito agradecemos.
