


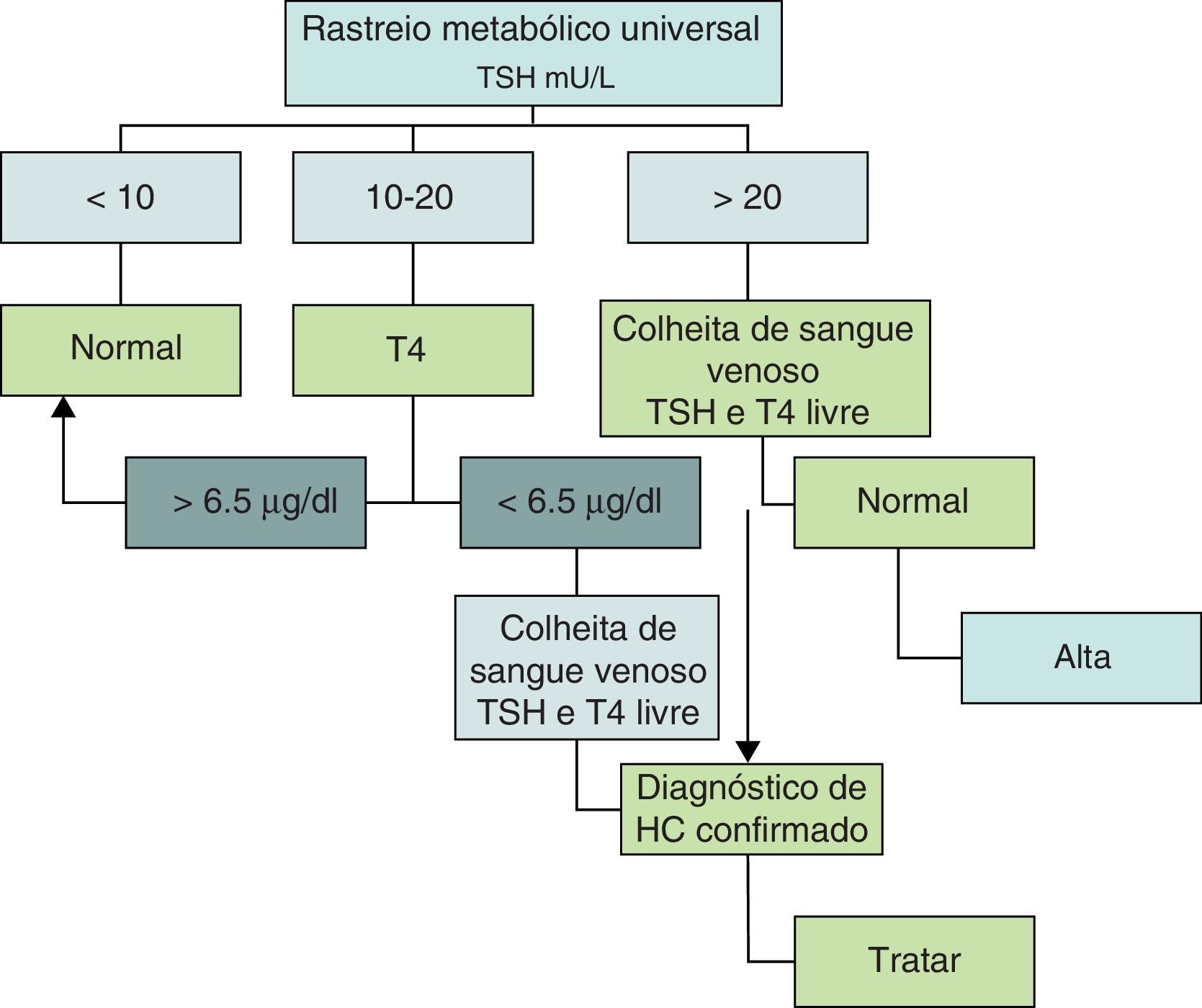

O hipotiroidismo congénito (HC) é a doença endócrina congénita mais frequente. Em Portugal, o rastreio universal teve início em 1981/82, através do doseamento de TSH a todos os recém‐nascidos entre o 3.° e o 6.° dias de vida. Valores de TSH inferiores a 10mU/L são considerados normais e entre 10‐20mU/L são considerados suspeitos, pelo que exigem a determinação da T4 na mesma amostra; se a T4 for inferior a 6,5μg/dl confirma‐se a suspeita de HC e o recém‐nascido deverá ser referenciado.
ObjetivoPretendeu‐se realizar uma revisão bibliográfica no sentido de apurar o estado da arte, nomeadamente em relação às atitudes de diagnóstico e terapêutica da doença. Propomos um esquema de follow‐up destes doentes que permita uma abordagem mais uniforme na prática clínica.
MétodosPesquisa sistemática nas bases de dados eletrónicas Medline e Pubmed com a palavra‐chave congenital hypothyroidism, incluindo estudos publicados até dezembro de 2012.
ConclusãoAs mutações genéticas responsáveis pelo HC dividem‐se em 2 grandes grupos: a grande maioria (85‐90%) causam alterações estruturais da tiróide, designando‐se no seu conjunto por disgenesia tiroideia e são maioritariamente esporádicas; as restantes, correspondendo a 10‐15% dos casos, causam alterações funcionais no processo de síntese das hormonas tiroideias (hormonogenese) e são transmitidas de forma autossómica recessiva.
A suspeita de HC deve ser confirmada através da avaliação da função tiroideia pelos métodos convencionais. A terapêutica com levotiroxina deve ser iniciada o mais precocemente possível, com o objetivo de normalizar a T4 em duas semanas e a TSH em quatro semanas. O seguimento dos recém‐nascidos com HC deve ser feito de 3 em 3 meses durante os primeiros 3 anos de vida.
Em suma, o diagnóstico precoce e tratamento imediato são de extrema importância evitando‐se desta forma as sequelas cognitivas graves do HC.
Congenital hypothyroidism (CH) is the most frequent endocrine congenital disorder. In Portugal, the universal screening was started in 1981/82, by TSH assay performed between the 3th and 6th day of life. Values under 10mU/L are considered normal, and between 10‐20 mU/L are suspicious, and it is necessary to determine T4 in the same sample. If T4 is less than 6,5μg/dl, the suspicion is confirmed and the newborn is then recalled.
PurposeTo review the related literature in order to establish the state of the art of CH, particularly in relation to diagnosis and treatment approach. We propose a follow up scheme for these patients to allow a more uniform approach to clinical practice.
MethodologySystematic research in the electronic databases Medline and Pubmed with the keyword: congenital hypothyroidism, including studies published up to December 2012.
ConclusionGenetic mutations responsible for CH are divided in two major groups. One group accounting for 85‐90% of the cases is related to structural changes of the thyroid gland, known under the common designation of thyroid dysgenesis and occurring mainly sporadically; the other group of patients with CH suffer of functional changes in the synthesis of the thyroid hormones (hormonogenesis), which altogether represents 10‐15% of the cases and are transmitted in an autosomal recessive manner.
The diagnosis should be confirmed by conventional thyroid function tests. Levothyroxine must be started as soon as possible, with the goal of achieving normal T4 in two weeks and normal TSH in one month. The follow‐up should be done at regular intervals of three months during the first 3 years of life.
In conclusion, it is extremely important to diagnose CH during the first days of live. Treatment should be started immediately in order to avoid an impairment of the cognitive development.
As hormonas tiroideias desempenham um papel fundamental no desenvolvimento e maturação do sistema nervoso central desde os seus estádios mais precoces, isto é, ainda durante a vida fetal e ao longo de toda a infância até a idade adulta1,2. A carência de hormonas tiroideias, particularmente nas formas severas de hipotiroidismo congénito (HC), pode ter consequências severas no desenvolvimento intelectual das crianças3,4. O diagnóstico precoce e o tratamento com levotiroxina permitem evitar as sequelas graves da doença, em particular as suas repercussões neurológicas5. Na década de 60, Robert Guthrie desenvolveu um método laboratorial fácil e acessível para medição da fenilalanina numa amostra de sangue total colhida em papel de filtro6. Este método conhecido inicialmente por «teste de Guthrie» foi posteriormente popularizado sob a designação de «teste do pezinho» atendendo a que o sangue é obtido através da punção cutânea do calcanhar. Começou por ser utilizado no rastreio da fenilcetonúria, mas poucos anos depois foi também adaptado ao rastreio do HC, que é cerca de 3‐4 vezes mais frequente do que a fenilcetonúria7. Ao longo da década de 70 a maioria dos países ocidentais adotou esta metodologia no rastreio universal dos recém‐nascidos8,9. Em 1982 todo o território nacional estava coberto pelo programa de rastreio do HC10,11. O sucesso obtido na erradicação do atraso mental grave, também conhecido por «cretinismo», contribuiu para que hoje o «teste do pezinho» seja utilizado no rastreio, não só do HC e da fenilcetonúria, mas também de outras 3 dezenas de doenças congénitas endócrinas e metabólicas.
Num levantamento por nós efetuado dos casos de HC diagnosticados nos últimos 30 anos nas ilhas de São Miguel e Santa Maria (Açores), obteve‐se uma incidência de 1:3420 recém‐nascidos, sobreponível aos dados nacionais. Estudos recentes apontam para um aumento na incidência mundial (1:2000‐1:3000 recém nascidos).
Ainda durante o curso deste levantamento casuístico foi identificada uma família com 3 gerações afetadas por HC. O estudo genético revelou uma nova mutação do gene que codifica o fator de transcrição PAX8 (c.74C>G) (Paired box 8). Para além do HC, os doentes afetados por esta mutação apresentavam malformações genitourinárias (Thyroid, in press)
A controvérsia sobre a metodologia do rastreio neonatal do HC, a investigação da sua etiologia e o estudo das malformações associadas exigem um conhecimento profundo dos mecanismos fisiopatológicos subjacentes à doença. Outra questão também pertinente é sobre o seguimento destes doentes: qual o intervalo mais apropriado? Que exames pedir e quando?
Com este trabalho pretende‐se uma revisão atualizada dos conhecimentos sobre HC, pelo que se procedeu a uma pesquisa bibliográfica sobre o tema e que serviu de base à elaboração do presente artigo.
MetodologiaFoi realizada uma pesquisa sistemática da literatura, utilizando‐se para o efeito as bases de dados eletrónicas Medline e PubMed. Os critérios de procura incluíram os estudos publicados até dezembro de 2012 obtidos através das palavras‐chave: congenital hypothyroidism. Só foram tomados em consideração os artigos em português ou inglês aos quais foi possível um acesso integral. Só em casos excecionais foram utilizados sites de referência, cuja informação não estava disponível sob outra forma.
Critérios de diagnósticoA estratégia de diagnóstico do HC, incluindo os parâmetros utilizados e os seus valores de referência, têm‐se alterado ao longo dos anos. Em quase todos os países da Europa, no Japão, Canadá e em parte dos Estados Unidos utiliza‐se a determinação do Thyroid Stimulating Hormone (TSH) como parâmetro inicial e o T4 (tiroxina) em segunda linha. Em outros países, particularmente na Holanda, o rastreio do HC tem por base a determinação inicial de T4 com a do TSH em segunda linha. Esta última abordagem tem a vantagem de poder diagnosticar os HC de causa central que cursam com hipotiroxinemia e TSH normal ou baixo. Apesar das discussões suscitadas, os diferentes algoritmos parecem equivaler‐se em termos de sensibilidade e especificidade, e ambos têm uma percentagem de referenciação de 0,05%, isto é, aproximadamente um falso positivo por cada caso diagnosticado12. Inicialmente a colheita de sangue era efetuada entre o 6.° e o 10.° dias de vida, mas como a permanência das puérperas no hospital é cada vez mais curta, a colheita tem vindo a ser efetuada mais precocemente, isto é, entre o3.° e 6.° dia de vida e ocasionalmente mais cedo, o que tem aumentado o número de falsos positivos13,14. Nunca é de mais referir que os falsos positivos representam um esforço suplementar no número de testes efetuados, bem como um motivo de preocupação para os pais, pelo que a colheita da amostra deve ser sempre realizada nas melhores condições e no intervalo de tempo recomendado. A necessidade de antecipar a data da colheita para o intervalo entre o 3.° e o 6.° dia está também relacionada com o facto de estar demonstrado que quanto mais precoce é o início da terapêutica menor é o risco de sequelas da doença14. No nosso país o valor limite do TSH era de 40mU/L até 2005, ano em que foi atualizado para os atuais 20mU/L15. Acima deste valor de TSH a suspeita de HC é elevada e o recém‐nascido é imediatamente referenciado. Por sua vez, valores de TSH abaixo de 10mU/L são considerados normais. Um TSH entre 10‐20mU/L é considerado suspeito e exige a determinação da T4 total na mesma amostra. Se o T4 for inferior a 6,5μg/dl confirma‐se a suspeita de hipotiroidismo e o recém‐nascido é então referenciado (fig. 1).
Exames complementares.")
Perante a suspeita de HC o recém‐nascido deve ser referenciado a uma consulta de endocrinologia pediátrica.
A primeira atitude é obter uma amostra de sangue venoso e avaliar a função tiroideia pelos métodos convencionais (TSH e T4 livre). Uma vez confirmada a suspeita, o tratamento com levotiroxina deve ser instituído de imediato, tendo em conta que a precocidade no início da terapêutica tem um impacto importante no prognóstico da doença. É neste contexto que a investigação da etiologia do HC deve ser sempre que possível adiada.
Não menos importante é a necessidade de proteger o recém‐nascido da agressividade dos meios complementares de diagnóstico. No entanto, estes pressupostos não invalidam a obtenção de uma anamnese cuidada e a realização de um exame objetivo pormenorizado. Uma história de consanguinidade paterna pode alertar‐nos para um defeito de dishormonogénese, cuja transmissão é quase sempre recessiva.
Por sua vez, a existência de múltiplos casos na família é a favor de uma situação de disgenesia da tiroideia. Uma história materna de doença de Graves alerta‐nos para a possibilidade de um HC transitório provocado por anticorpos que atingem o feto por via transplacentar e que bloqueiam o recetor TSH fetal. No exame objetivo podemos estar perante um recém‐nascido com poucos ou nenhuns sinais de hipotiroidismo ou pelo contrário, apresentar‐se ictérico, com macroglossia, hérnia umbilical e choro rouco, evidenciando um quadro clínico de HC grave. A presença de bócio, particularmente quando volumoso, é a favor de dishormonogénese. A existência de fenda palatina está associada a situações de disgenesia.
Entre os exames complementares a ecografia ocupa um lugar privilegiado. É um exame inócuo e facilmente acessível, tem ganho uma maior precisão com a utilização simultânea do doppler16,17. Permite definir com maior exatidão a localização, bem como as dimensões e características da tiroideia, nem sempre possíveis de avaliar pela simples palpação, em particular no recém‐nascido. A ausência de tecido tiroideu na sua localização habitual é sugestiva de atireose ou de ectopia, uma situação por vezes só possível de diferenciar pela cintigrafia. Mas a ecografia permite ainda confirmar ou excluir a existência de malformações renais ou cardíacas, frequentemente associadas a situações de disgenesia.
Sempre que a cintigrafia da tiroideia se torne imprescindível na avaliação da etiologia do HC, a sua realização é habitualmente realizada após os 2 anos de idade. Em primeiro lugar porque exige na maioria das situações a suspensão temporária da terapêutica com levotiroxina. Em segundo lugar, porque requer uma maior participação do doente. Finalmente, não sendo um exame acessível em todos os centros, a sua realização obriga a deslocações prolongadas sempre difíceis para um recém‐nascido. As informações obtidas pela cintigrafia são não só morfológicas, mas também de carácter funcional. Permitem o diagnóstico diferencial definitivo entre a atireose e a ectopia da glândula. Quando associada ao teste do perclorato pode dar‐nos informações sobre os défices de organificação do iodo.
De salientar a importância do doseamento da tiroglobulina (TG) que em conjunto com os achados ecográficos pode desempenhar um papel importante no estudo da etiologia do HC. A ausência de glândula tiroideia na sua topografia habitual em simultâneo com níveis baixos ou indoseáveis de TG apontam para uma situação de agenesia, tornando assim pouco provável a possibilidade de ectopia ou hipoplasia da glândula. Por sua vez, na presença de uma glândula normal ou aumentada associada a níveis circulantes de TG baixos ou indoseáveis, os defeitos da síntese de TG são a hipótese mais provável. Finalmente, valores elevados de TG ocorrem em situações de dishormonogénese.
Nunca é demais salientar que mais de 80% dos casos de HC ocorrem de forma esporádica. Nestas circunstâncias a investigação dos defeitos genéticos subjacentes à doença é completamente inconsequente. Os testes genéticos devem ser solicitados em situações em que a incidência familiar da doença é particularmente evidente. A identificação de uma mutação genética associada à transmissão da doença pode ter um papel importante no aconselhamento dos doentes e seus familiares. Pode ainda condicionar a abordagem terapêutica. De facto e como adiante discutiremos, existem circunstâncias em que a simples suplementação de iodo pode ser suficiente para corrigir algumas formas hereditárias de HC.
Causas de hipotiroidismo congénitoNuma perspetiva global, a causa mais frequente de hipotiroidismo, incluindo a sua forma congénita, é a carência de iodo que atinge cerca de 1/3 da população mundial18. A diminuição do aporte de iodo, particularmente durante a gravidez, impede a formação de hormonas tiroideias em quantidades adequadas para suprir as necessidades acrescidas da unidade feto‐materna. Mesmo que o défice hormonal seja modesto as suas consequências neurológicas não são desprezíveis e preocupam de sobremaneira as autoridades sanitárias em todo o mundo. Por sua vez, o HC na sua variante não endémica atinge 1:3.000/4.000 nados vivos19, sendo por isso a doença endócrina congénita mais comum. As alterações do desenvolvimento embrionário da tiroideia (agenesia, ectopia ou hipoplasia), designadas no seu conjunto por disgenesia, são responsáveis por cerca de 85‐90% dos casos20. Nos restantes 10‐15%, a insuficiência de produção de hormonas tiroideias está relacionada com defeitos enzimáticos que afetam direta ou indiretamente a disponibilidade intracelular de iodo, bem como a sua organificação, eventos cruciais no processo de síntese das hormonas tiroideias (hormonogenese)21. Os defeitos da hormonogenese cursam habitualmente com glândulas tiroideias aumentadas de volume e que podem atingir dimensões consideráveis se não forem tratados a tempo. Mais raramente, em cerca de 1:10.000 ou 20.000 nados vivos a causa do HC é de origem central, isto é, por doença hipofisária ou hipotalâmica (hipotiroidismo secundário e terciário, respetivamente) e coexistem com uma tiroide estrutural e funcionalmente íntegra22,23. Estas formas de HC, que são muitas vezes transitórias, decorrem de uma deficiente estimulação da glândula tiroideia por uma produção inadequada de TSH muitas vezes associada à imaturidade do eixo hipotálamo‐hipófise‐tiroideia como por vezes acontece nos recém‐nascidos prematuros.
A disgenesia da tiroideia é tradicionalmente considerada uma doença esporádica, com uma incidência cerca de 2 vezes superior no sexo feminino. De facto, cerca de 90% dos gémeos homozigóticos são discordantes em relação à doença24 e em apenas 2% dos casos foi demonstrada a coexistência da doença em familiares do primeiro grau25. No seu conjunto, estes achados parecem constituir argumentos suficientes para excluir uma transmissão genética segundo um modelo mendeliano clássico. No entanto, cerca de 7% destes doentes apresentam anomalias congénitas extratiroideias, uma prevalência elevada quando comparada com a observada na população em geral, que não ultrapassa os 2,5%26,27. Para além disto, cerca de 1,5% dos casos de HC são também portadores de pequenas aberrações cromossómicas28,29. Finalmente, estudos populacionais recentes demonstraram que a probabilidade de ocorrência de disgenesia em familiares em primeiro grau é cerca de 15 vezes maior do que aquela que seria esperada se a sua distribuição fosse aleatória30. Numa tentativa de integrar estas observações aparentemente contraditórias, várias hipóteses têm sido formuladas. Em primeiro lugar, admite‐se que a existência de mutações pós‐zigóticas epigenéticas possam, por si só, justificar uma parte considerável dos casos da doença31,32. Por sua vez, um modelo de «dupla agressão» permitiria conciliar alguns aspetos contraditórios como a discordância entre gémeos homozigóticos e uma frequência maior do que a esperada em familiares de primeiro grau33. Neste modelo patogénico e tal como tem sido admitido no retinoblastoma ou no MEN1, o primeiro golpe seria uma alteração de natureza genética, enquanto o segundo golpe resultaria da ação mutagénica de fatores extrínsecos capaz de inativar o locus contralateral (homozigotia) ou um outro locus, criando assim uma dupla heterozigotia (heterozigóticos compostos). Parece‐nos assim admissível que na etiologia da disgenesia tiroideia possam participar fatores genéticos de carácter mendeliano, isto é, inscritos no DNA e fatores epigenéticos, provavelmente modulados pelo género e por outras circunstâncias intrínsecas ou extrínsecas. Pelo contrário, na maioria dos casos de dishormonogénese tem sido possível estabelecer um padrão de transmissão autossómico recessivo21.
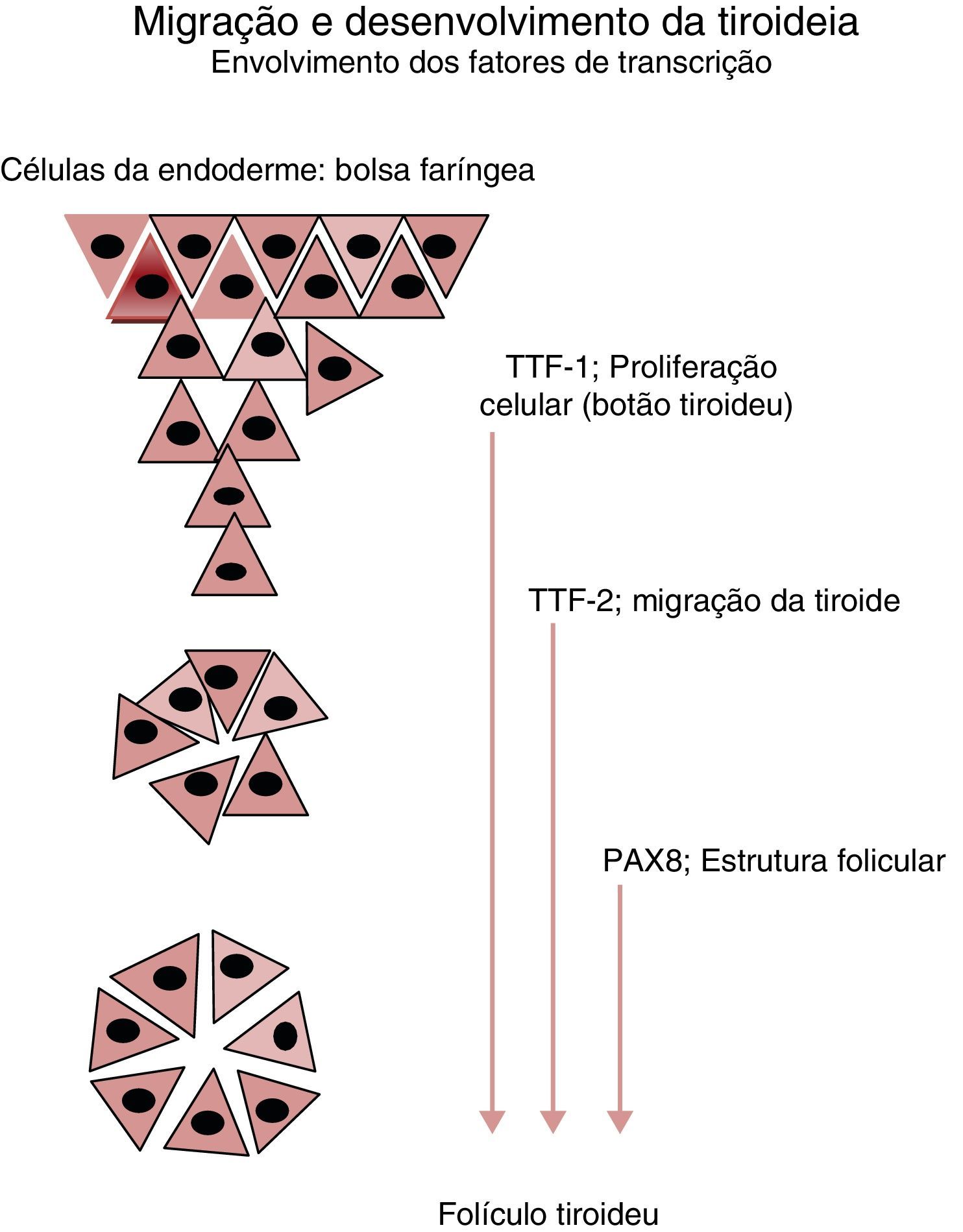
Nos cerca de 2‐3% dos casos em que a ocorrência familiar da disgenesia obedece a um padrão mendeliano clássico, a identificação dos genes envolvidos na transmissão da doença tem permitido clarificar aspetos importantes no desenvolvimento embrionário da glândula, particularmente na sua migração desde a base da língua até à sua localização habitual (fig. 2), bem como maturação funcional da glândula ao longo da gravidez (fig. 3). Da mesma forma, a caracterização molecular das mutações causadoras de dishormogenese tem contribuído para o esclarecimento de muitos aspetos funcionais da glândula tiroideia, que por sua vez permitiram uma melhor compreensão dos mecanismos fisiopatológicos responsáveis pelo HC e pelas doenças da tiroideia em geral.
 participa nos estadios iniciais da organogénese da glândula tiroideia, inibindo a apoptose e controlando a sobrevivência das células percussoras. O Thyroid Transcription Factor 2 (TTF‐2) promove a migração destas células. O Paired box 8 gene (PAX8) possui um papel fundamental na cascata reguladora da diferenciação funcional das células foliculares tiroideias.")
Principais genes envolvidos no desenvolvimento e migração da glândula tiroideia. A partir do estudo de doentes afetados por mutações destes genes de fatores de transcrição foi possível estabelecer a sua influência em cada um dos eventos representados. O Thyroid Transcription Factor 1 (TTF‐1) participa nos estadios iniciais da organogénese da glândula tiroideia, inibindo a apoptose e controlando a sobrevivência das células percussoras. O Thyroid Transcription Factor 2 (TTF‐2) promove a migração destas células. O Paired box 8 gene (PAX8) possui um papel fundamental na cascata reguladora da diferenciação funcional das células foliculares tiroideias.
.")
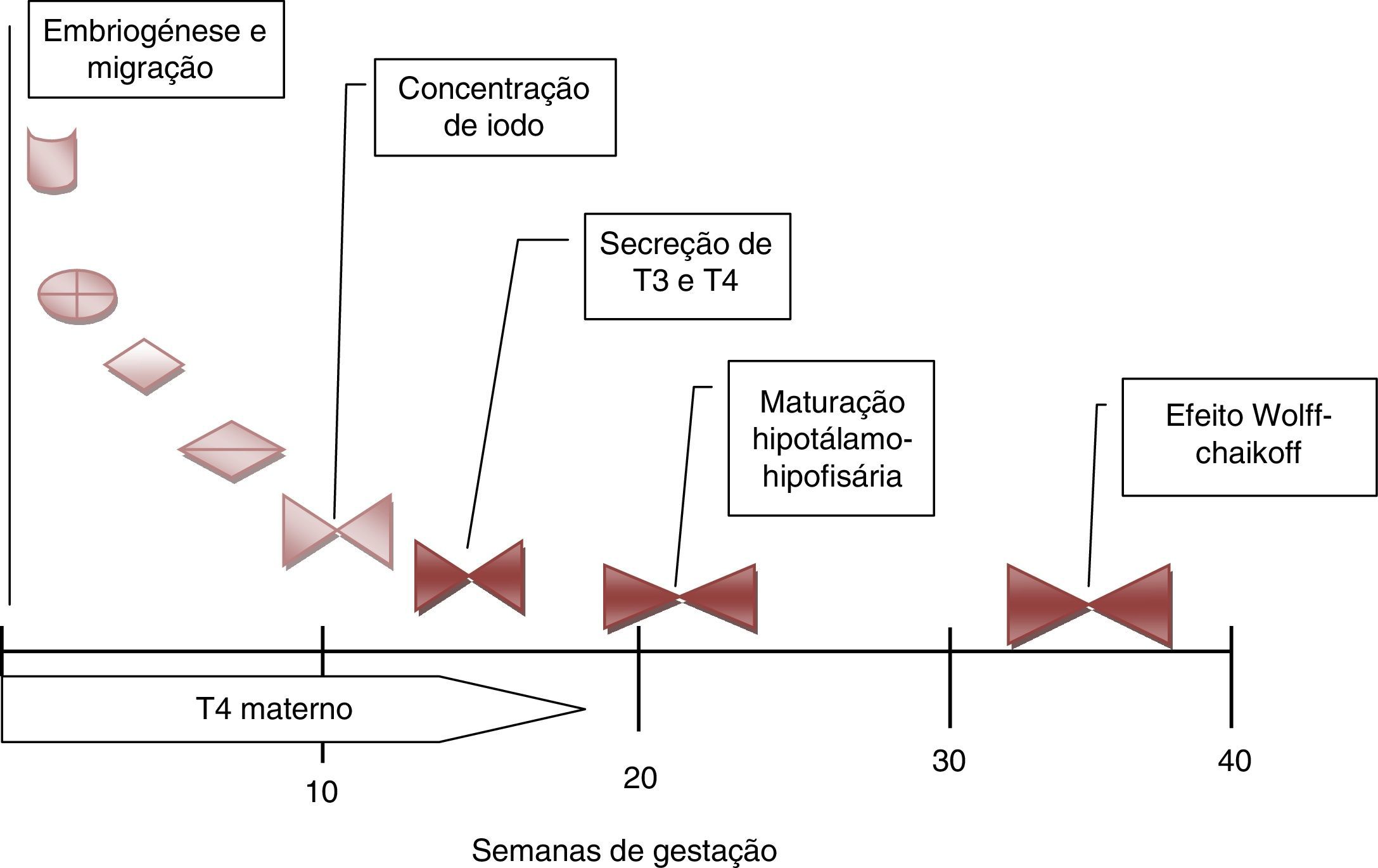
Desenvolvimento e maturação da tiroideia fetal durante a gravidez. De salientar a importância das hormonas tiroideias maternas durante o primeiro trimestre da gravidez. A maturação do eixo hipotálamo‐hipófise‐tiroideia só está completa por volta das 20 semanas. Só muito tardiamente a glândula tiroideia fetal é afetada pela sobrecarga de iodo (T4: tiroxina; T3: triiodotiroxina).
As mutações genéticas responsáveis pelo HC dividem‐se assim em 2 grandes grupos:
- 1)
As que produzem alterações estruturais da glândula tiroideia e que no seu conjunto se designam por disgenesia da tiroideia (agenesia, ectopia ou hipoplasia): (tabela 1)
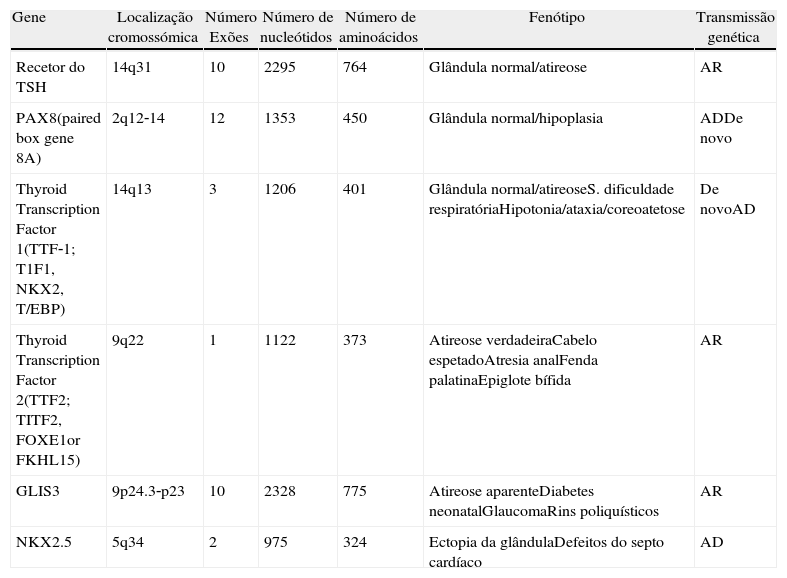
Tabela 1.Causas monogénicas de disgenesia da tiroideia
Gene Localização cromossómica Número Exões Número de nucleótidos Número de aminoácidos Fenótipo Transmissão genética Recetor do TSH 14q31 10 2295 764 Glândula normal/atireose AR PAX8(paired box gene 8A) 2q12‐14 12 1353 450 Glândula normal/hipoplasia ADDe novo Thyroid Transcription Factor 1(TTF‐1; T1F1, NKX2, T/EBP) 14q13 3 1206 401 Glândula normal/atireoseS. dificuldade respiratóriaHipotonia/ataxia/coreoatetose De novoAD Thyroid Transcription Factor 2(TTF2; TITF2, FOXE1or FKHL15) 9q22 1 1122 373 Atireose verdadeiraCabelo espetadoAtresia analFenda palatinaEpiglote bífida AR GLIS3 9p24.3‐p23 10 2328 775 Atireose aparenteDiabetes neonatalGlaucomaRins poliquísticos AR NKX2.5 5q34 2 975 324 Ectopia da glândulaDefeitos do septo cardíaco AD - 2)
As que causam alterações funcionais no processo de síntese das hormonas tiroideias nomeadamente na organificação do iodo (hormonogenese): (tabela 2).
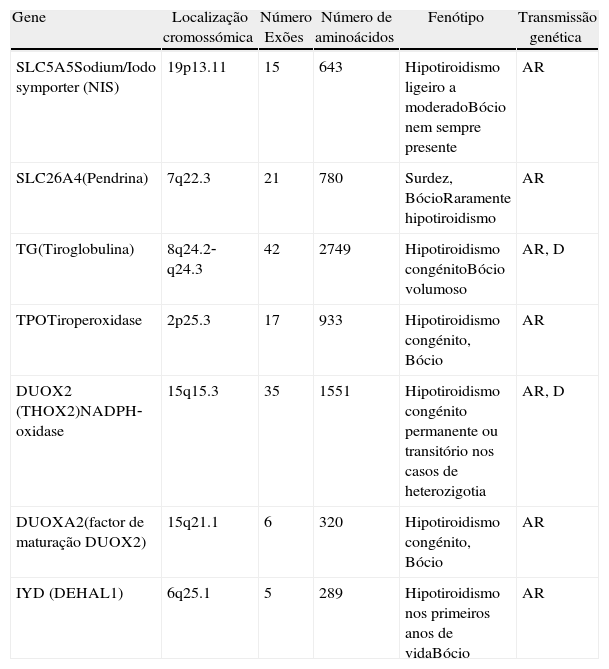
Tabela 2.Causas monogénicas de dishormonogénese
Gene Localização cromossómica Número Exões Número de aminoácidos Fenótipo Transmissão genética SLC5A5Sodium/Iodo symporter (NIS) 19p13.11 15 643 Hipotiroidismo ligeiro a moderadoBócio nem sempre presente AR SLC26A4(Pendrina) 7q22.3 21 780 Surdez, BócioRaramente hipotiroidismo AR TG(Tiroglobulina) 8q24.2‐q24.3 42 2749 Hipotiroidismo congénitoBócio volumoso AR, D TPOTiroperoxidase 2p25.3 17 933 Hipotiroidismo congénito, Bócio AR DUOX2 (THOX2)NADPH‐oxidase 15q15.3 35 1551 Hipotiroidismo congénito permanente ou transitório nos casos de heterozigotia AR, D DUOXA2(factor de maturação DUOX2) 15q21.1 6 320 Hipotiroidismo congénito, Bócio AR IYD (DEHAL1) 6q25.1 5 289 Hipotiroidismo nos primeiros anos de vidaBócio AR
Fazem parte deste grupo os genes que codificam o recetor do TSH e 3 fatores de transcrição, a saber: Thyroid Transcription Factor 1 (TTF‐1); Thyroid Transcription Factor 2 (TTF‐2) e o fator de transcrição denominado PAX8.
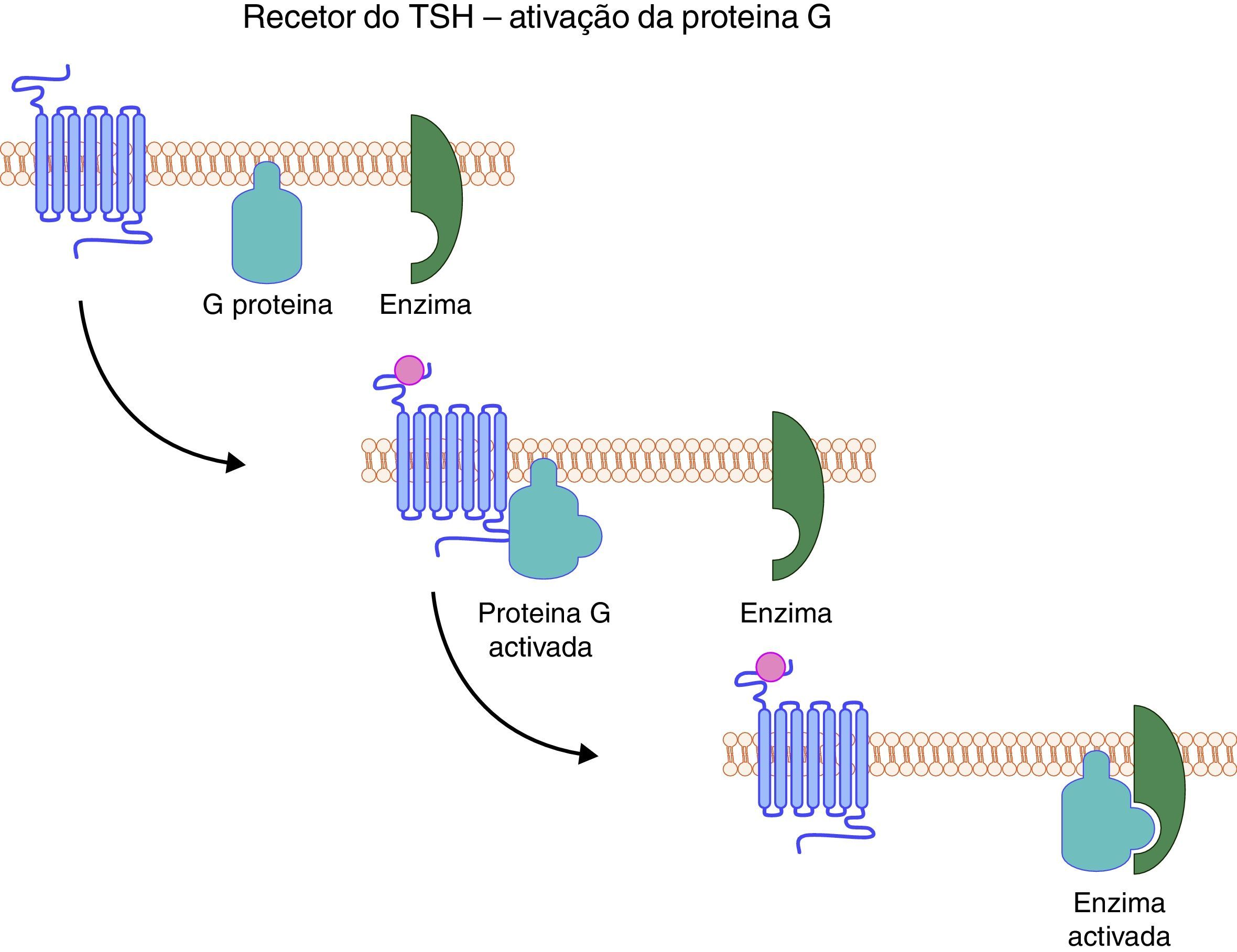
O gene do recetor de TSH codifica uma proteína que ocupa a superfície das células foliculares da tiroideia, mergulhando no interior da membrana até ao citoplasma através de uma estrutura helicoidal. Esta estrutura é responsável não só pela ancoragem da proteína, mas também pela transmissão do sinal da superfície celular para o citoplasma. Do ponto vista estrutural, o recetor do TSH pertence a uma subfamília de proteínas G que têm em comum a mesma estrutura, isto é, 7 segmentos transmembrana, com 3 ansas intracelulares e 3 extracelulares. O seu domínio extracelular é codificado por 9 exões, enquanto a porção embebida na membrana e a porção intracelular são codificadas apenas por um único exão. A sequência de eventos despoletada pela ligação do TSH ao seu recetor culmina com o aumento dos níveis de AMPc intracelular, bem como de outros mediadores como GMPc e o fosfolípido C (fig. 4). A primeira família com uma mutação do gene do recetor de TSH foi identificada em 199534. Desde então múltiplas mutações homozigóticas ou heterozigóticas compostas têm sido descritas em famílias que apresentam fenótipos variáveis. De facto, a resistência ao TSH pode manifestar‐se sob a forma de uma elevação isolada da tirotrofina na presença de uma glândula tiroideia normal, até ao extremo oposto, isto é, sob a forma de um HC grave no contexto de uma glândula tiroideia atrófica ou apenas vestigial35–37.
.")
O recetor do TSH uma vez ativado liga‐se à proteína G, que por sua vez ativa a cascata enzimática que culmina com o aumento dos níveis intracelulares de AMPc, bem como de fosfolípidos. O TSH influencia não só a produção de hormonas tiroideias, mas também o crescimento e diferenciação da glândula (TSH: Thyroid Stimulating Hormone).
A participação dos fatores de transcrição na embriogénese e migração da glândula tiroideia é exercida fundamentalmente através da ativação dos genes da tiroglobulina, tiroperoxidase (TPO) e da proteína de transporte do iodo («sodium/iodo symporter»), bem como pela sua influência na diferenciação e manutenção das células foliculares da tiroideia, embora esta última ação ocorra através de um mecanismo ainda mal esclarecido38. Estão nestas circunstâncias os fatores de transcrição TTF‐1, TTF‐2 e o PAX8 (fig. 2).
O TTF‐1 faz parte de uma família de fatores de transcrição caracterizada pela existência de um domínio «homeobox» comum a todos os membros da família. É através deste domínio conservador que os fatores de transcrição desta família se ligam a elementos específicos do DNA39. Na glândula tiroideia o TTF‐1 regula a transcrição dos genes da TG e da TPO. Para além da tiroide, a sua expressão tem sido documentada nas células epiteliais do pulmão onde é responsável pela transcrição da proteína do surfatante, bem como no cérebro onde desempenha um importante papel no desenvolvimento do diencéfalo40. A sua mutação de novo ou transmitida de forma autossómica dominante está associada à síndrome de dificuldade respiratória neonatal por défice de surfatante pulmonar que em simultâneo com as manifestações neurológicas como a hipotonia, disartria, ataxia, coreoatetose e microcefalia fazem parte da trilogia «cérebro‐tiroide‐pulmão» característica das mutações deste gene41. Entre estes 3 componentes, as alterações da tiroideia são as menos graves. As lesões mais graves são as pulmonares que podem conduzir à morte por agenesia do pulmão. Por sua vez as manifestações neurológicas são habitualmente ligeiras a moderadas.
O NKX2.5 é outro fator de transcrição que pertence a uma superfamília de fatores de transcrição caraterizada pela presença de um domínio «homeobox» na estrutura tridimensional da proteína. Está sobretudo envolvido na embriogénese da tiroide e do miocárdio. Trata‐se assim de um forte candidato em casos de HC associado a defeitos cardíacos20.
O TTF‐2 é uma fosfolipoproteína pertencente a uma família de fatores de transcrição que inclui na sua estrutura um domínio em cabeça de garfo (ou hélix alada). É através deste domínio que o TTF‐2 se liga a sequências específicas do DNA, regulando a transcrição dos genes da TG e da TPO42. Até à presente data, são conhecidas apenas 3 mutações, todas elas homozigóticas. As mutações descritas são responsáveis por um quadro clínico florido no qual se incluem a agenesia da tiroideia, fenda palatina, epiglote bífida, cabelo eriçado e atrésia das coanas (síndrome de Bamforth‐Lazarus)43,44.
O PAX8 é um dos 9 membros de uma família de fatores de transcrição que incluem na sua estrutura um domínio emparelhado através do qual se ligam ao DNA45,46. A maioria das mutações até agora descritas situam‐se na região mais conservadora do domínio emparelhado e têm como consequência uma incapacidade de ligação do fator de transcrição ao DNA47. O PAX8 promove a transcrição dos genes da TPO, da TG e do transportador de sódio/iodo48. Diversos estudos têm demonstrado que a sua ação é imprescindível não só no início da diferenciação das células foliculares da tiroideia, mas também na sua manutenção e proliferação ao longo da vida49. Os indivíduos heterozigóticos para mutações do gene PAX8 apresentam habitualmente glândulas tiroideias eutópicas, de dimensões reduzidas ou normais. Em apenas um caso isolado foi sugerida a existência de uma glândula tiroideia ectópica num doente com mutação de PAX8, não tendo, contudo, sido confirmada por cintigrafia45. Em outro caso isolado, a glândula tiroideia era normal ao nascer, mas regrediu ao longo dos primeiros anos de vida, dando origem a uma forma de hipotiroidismo não autoimune com início tardio50. Em conclusão, as diferentes mutações do gene PAX8 até agora descritas estão associadas a manifestações clínicas muito variáveis, inclusive numa mesma família, sugerindo uma modulação do fenótipo por múltiplos fatores51. Todos os indivíduos com mutações PAX8 até agora descritos são heterozigóticos para a mutação52. Diversas hipóteses têm sido admitidas para justificar a transmissão autossómica dominante da doença em circunstâncias de heterozigotia. A possibilidade da existência de um mecanismo de interação negativa dominante parece pouco provável e foi apenas demonstrado numa mutação53. Estudos de cotransfecção da proteína mutante em simultâneo com a normal não demonstraram qualquer efeito negativo dominante nas restantes mutações já descritas. A expressão monoalélica do gene mutante, associada ou não a um fenómeno de imprinting em que subsistem 2 populações diferentes de células tiroideias, é outra alternativa para a qual as evidências laboratoriais existentes são contraditórias54. Finalmente, a hipoplasia da tiroideia nos doentes heterozigóticos para mutações PAX8 pode estar relacionada com um fenómeno de dosagem do gene, isto é, o desenvolvimento de uma glândula tiroideia normal, morfológica e funcional requer uma determinada concentração de PAX855.
A redução dos níveis de PAX8 por inativação de um dos alelos (haploinsuficiência) produz um desempenho funcional menos eficiente do PAX8 na transcrição dos genes alvo, incluindo os genes envolvidos na proliferação e diferenciação celular das células da tiroideia56.
A associação de mutações do PAX8 com agenesia renal unilateral tem sido raramente descrita52,57. A expressão da proteína PAX8 foi demonstrada em fases muito precoces do desenvolvimento embrionário da glândula tiroideia. O mesmo se tem verificado durante o desenvolvimento renal, o que sugere que o PAX8 pode ter um papel relevante na embriogénese renal, tal como o PAX258. Por sua vez, a expressão do PAX8 tem sido documentada em células de tumores de origem renal59,60. Este achado parece indicar que o processo de indiferenciação das células tumorais pode induzir a expressão de proteínas características das células embrionárias, constituindo assim mais uma evidência do papel do PAX8 na embriogénese renal.
As mutações no gene GLIS3 são uma causa rara de diabetes neonatal e HC. Estudos recentes demonstram uma grande variabilidade fenotípica, ocorrendo muitas vezes fibrose hepática, displasia poliquística renal, glaucoma, dismorfismos faciais e alterações esqueléticas. Os mecanismos fisiopatológicos envolvidos no hipotiroidismo ainda não estão bem esclarecidos, no entanto, parecem estar relacionados com uma resistência parcial ou completa ao TSH.
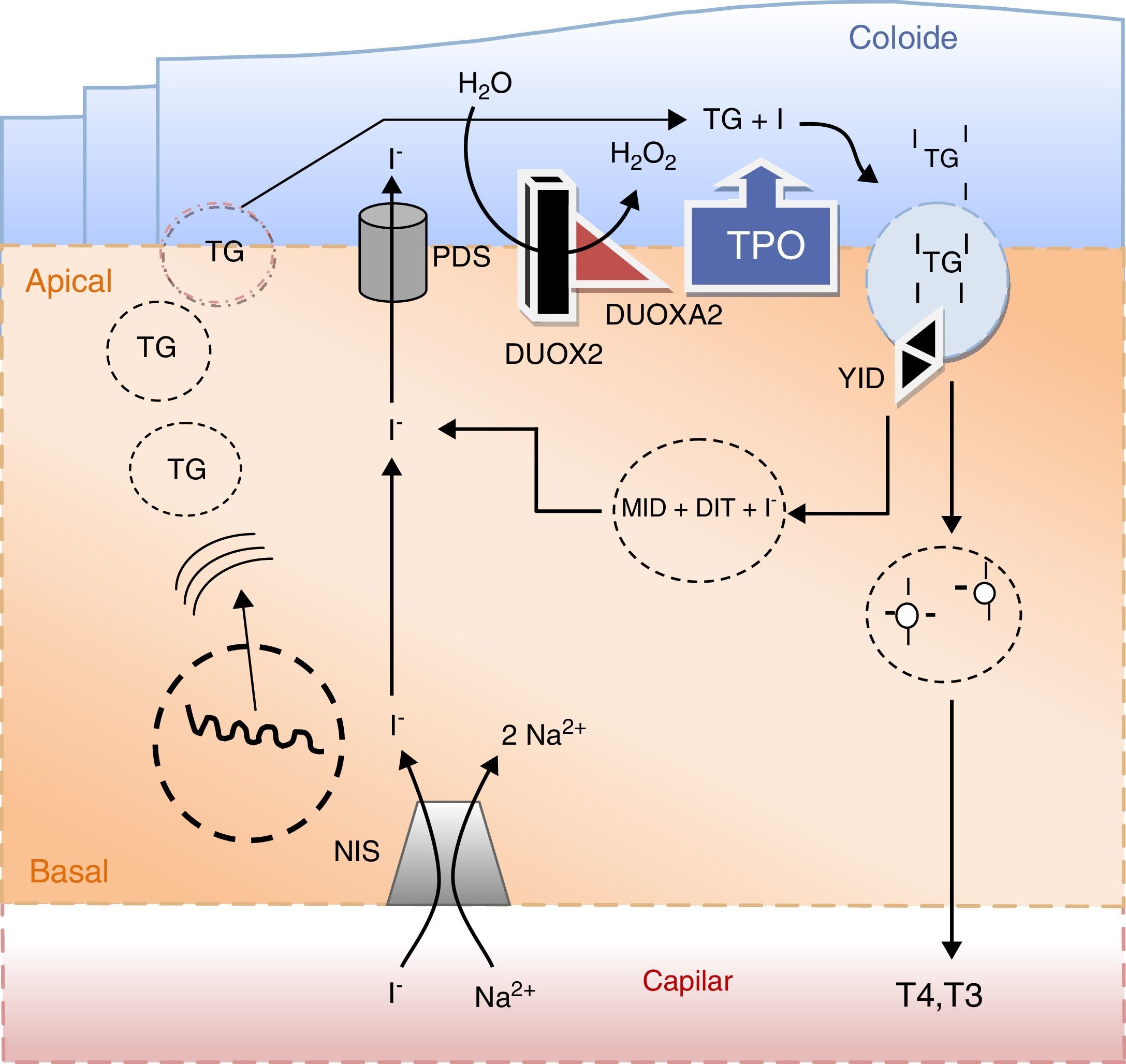
Causas monogénicas de dishormonogéneseA função do eixo hipotálamo‐hipofisário‐tiroideia é providenciar hormonas tiroideias em quantidades adequadas às necessidades do organismo. Este objetivo pressupõe a plena integridade e funcionalidade de todo o eixo, bem como a existência de matéria‐prima com especial relevância para o iodo. O processo designado por hormonogenese, tem início com o transporte do iodo para o interior das células foliculares da tiroideia e culmina com a libertação de hormonas ativas na circulação (fig. 5).
 é concentrado ativamente na célula folicular da tiroideia através do transportador sódio/iodo symporter (NIS) localizado na membrana baso‐lateral da célula. Uma vez no polo apical da célula, o iodeto passa para o coloide por um processo de difusão facilitada mediado pela pendrina (PDS). A enzima peroxidase (TPO) na presença de peroxido de hidrogénio (H2O2) gerado pelo sistema de NADPH‐oxidases (DUOX e DUOXA, dual oxidase maturation factors) cataliza a ligação covalente do iodo aos resíduos tirosil da tiroglobulina (TG) e a subsequente formação de mono e diiodotironinas (MIT e DIT). As enzimas lisossomais libertam as hormonas tiroideias ativas T4 (tiroxina) e T3 (triiodotiroxina) da sua matriz. O iodo remanescente é reciclado por ação da desalogenase (YID). A secreção das hormonas tiroideias ativas na circulação dá‐se através de um processo de exocitose.")
Principais eventos na síntese das hormonas tiroideias e no metabolismo do iodo. O iodo sob a forma de iodeto (I−) é concentrado ativamente na célula folicular da tiroideia através do transportador sódio/iodo symporter (NIS) localizado na membrana baso‐lateral da célula. Uma vez no polo apical da célula, o iodeto passa para o coloide por um processo de difusão facilitada mediado pela pendrina (PDS). A enzima peroxidase (TPO) na presença de peroxido de hidrogénio (H2O2) gerado pelo sistema de NADPH‐oxidases (DUOX e DUOXA, dual oxidase maturation factors) cataliza a ligação covalente do iodo aos resíduos tirosil da tiroglobulina (TG) e a subsequente formação de mono e diiodotironinas (MIT e DIT). As enzimas lisossomais libertam as hormonas tiroideias ativas T4 (tiroxina) e T3 (triiodotiroxina) da sua matriz. O iodo remanescente é reciclado por ação da desalogenase (YID). A secreção das hormonas tiroideias ativas na circulação dá‐se através de um processo de exocitose.
O iodo, sob a forma de iodeto, é transportado ativamente da corrente sanguínea para as células foliculares da tiroideia em simultâneo com o sódio na proporção de 1:2, isto é, I‐/2Na+. Este processo é mediado por um transportador localizado na membrana basal da célula folicular designado por sódio‐iodo «symporter» (NIS). O NIS é uma glicoproteína de 643 aminoácidos codificado pelo gene SLC5A561. O processo de transporte utiliza um gradiente eletroquímico gerado pelas Na/K‐ATPases. A expressão do transportador é modulada pelo TSH e pelo iodo agindo quer a nível da transcrição do gene quer sobre a sua translação62. O TSH via AMPc estimula a expressão de NIS, não estando contudo documentada uma ação reguladora sobre a sua atividade. Por sua vez, o iodo tem um efeito inibitório conhecido por efeito Wolff‐Chaikoff que regula a disponibilidade intracelular deste oligoelemento em situações de excesso de oferta. O mecanismo subjacente é complexo e mediado por múltiplos genes, bem como por diversas proteínas existentes no citoplasma da célula folicular63,64. Este efeito inibitório é, no entanto, autolimitado e dá lugar ao chamado fenómeno de escape, 2‐3 semanas após o início da sobrecarga iodada. Apesar de ter uma elevada afinidade para o iodo, o NIS pode também transportar outros iões. Catiões de grandes dimensões como o perclorato inibem a concentração de iodo na tiroideia uma vez que competem para o mesmo transportador65. Tendo em conta este efeito, o perclorato bem como os tiocianatos foram utilizados no tratamento do hipertiroidismo, mas posteriormente abandonados em virtude da sua elevada toxicidade. O perclorato continua, no entanto, a ser utilizado em provas funcionais. Na presença de um defeito de organificação de iodo, uma parte substancial do iodo radioativo administrado por via oral ou intravenosa permanece livre na glândula tiroideia. A administração subsequente de perclorato origina uma descarga na corrente sanguínea do iodo não organificado com a consequente diminuição da radioatividade da glândula. Numa tiroideia normal a radioatividade mantém‐se estável uma vez que grande parte do iodo administrado é imediatamente organificado. Este é o fundamento do chamado teste de descarga do perclorato usado na avaliação dos défices de organificação do iodo66.
A presença de NIS tem sido documentada em muitos outros órgãos para além da tiroideia nomeadamente nas glândulas salivares e na mucosa gástrica, mas a sua existência na glândula mamária é particularmente relevante, uma vez que permite a concentração de iodo no leite materno disponibilizando ao recém‐nascido um suplemento adequado deste oligoelemento62. A primeira mutação homozigótica deste gene foi descrita em 197761. Desde então têm sido descritas muitas outras mutações transmitidas de uma forma autossómica recessiva. Todas elas cursam habitualmente com bócio e hipotiroidismo cuja gravidade é muito variável e nem sempre evidente. A manifestação mais característica nos doentes com mutações deste gene, é a ausência de captação de iodo radioativo na gamagrafia da tiroideia, semelhante ao que acontece na atireose e com a qual pode ser confundida. A existência de um quociente baixo, próximo da unidade, na relação iodo na saliva versus no plasma é outra alteração característica da doença67. Em muitos casos de hipotiroidismo provocados pela mutação do gene SLC5A5 a suplementação de iodo pode ser suficiente para a normalização da função tiroideia, pelo que quer o bócio quer o hipotiroidismo são mais comuns nas regiões com carência de iodo68.
Quando atinge a zona apical da célula folicular, o iodo ainda sob a forma de iodeto é exportado de forma passiva para o folículo tiroideu através de uma proteína de transporte conhecida por pendrina (PDS). A PDS é uma glicoproteína com 780 aminoácidos codificada pelo gene SLC26A469. Funciona como transportador de iodeto na membrana apical da célula folicular, bem como no ouvido interno e rim. Neste último órgão tem um papel relevante na regulação do metabolismo ácido/base intervindo nas trocas de cloro e bicarbonato70. A PDS desempenha também um papel importante na cóclea assegurando a manutenção do pH da endolinfa que é o veículo de transmissão dos potenciais endococleares gerados pelos estímulos sonoros71. As mutações do gene SLC26A4 cuja incidência, tendo por base estudos efetuados na Inglaterra, é de 1:600.000 nados vivos, originam a síndrome de Pendred descrita pela primeira vez em 1896. A doença é transmitida de forma autossómica recessiva e caracteriza‐se pela coexistência de surdez neurossensorial e bócio. Só muito raramente estes doentes são detetados pelo rastreio neonatal do HC, sendo a surdez a principal manifestação da doença e aquela que em primeiro lugar chama a atenção médica72. De facto, cerca de 10% dos casos de surdez congénita são provocados por mutações da PDS que são no seu conjunto responsáveis por cerca de 80% dos casos de surdez sindromática, isto é, surdez congénita associada a outras alterações morfológicas ou funcionais, nomeadamente da tiroideia73. Caracteristicamente estes doentes exibem uma dilatação do ducto e do saco endolinfático visível nos exames morfológicos, nomeadamente na ressonância magnética. O chamado defeito de Mondini que consiste na substituição dos canais cocleares por uma cavidade única é menos comum74,75. O bócio manifesta‐se habitualmente durante a segunda década de vida e pode ser difuso ou nodular. Apesar de apresentarem níveis de TSH no limite superior do normal, raramente estes doentes evoluem para hipotiroidismo, que tal como o bócio é mais frequente nas regiões com carência de iodo. Este efeito modulador do iodo na expressão fenotípica da doença é a explicação mais plausível para o facto de cerca de 50% das mutações do gene SLC26A4 até agora descritas não apresentarem alterações da tiroideia, manifestando‐se apenas por surdez (surdez congénita não sindromática)76.
O constituinte mais abundante no coloide é a TG, uma proteína extensa, das mais extensas do organismo humano, sintetizada nos ribossomas da célula folicular sob a forma de uma pré‐molécula com 2.749 aminoácidos e que atinge os ácinos através de um processo de exocitose77. Cada molécula de TG contém cerca de 120 resíduos de tirosina que uma vez organificados dão origem a 4 a 6 moléculas de hormonas tiroideias, pelo que podemos considerar a TG como um molde no processo de síntese das hormonas tiroideias78.
As mutações deste gene manifestam‐se por HC com bócios habitualmente volumosos e com níveis circulantes baixos ou indoseáveis de tiroglobulina78. Uma particularidade destes doentes é apresentarem níveis de T3 livre proporcionalmente mais elevados que os de T4 livre, o que supostamente depende de uma maior atividade intratiroideia da desiodase tipo 279,80. A imagem cintigráfica da tiroideia é habitualmente normal uma vez que o processo de organificação do iodo pode, em alternativa à tiroglobulina, utilizar outras proteínas intracelulares ou intrafoliculares, como por exemplo a albumina. As alterações da estrutura tridimensional da molécula provocadas pelas mutações do gene da TG, particularmente aquelas que afetam os resíduos de cisteína, originam a sua retenção sob a forma de agregados no retículo endoplasmático da célula folicular da tiroideia e que do ponto de vista morfológico são em tudo semelhantes a uma doença de acumulação do retículo endoplasmático81–83.
A TPO é uma hemo‐proteína glicosilada com 933 aminoácidos ancorada na membrana apical das células foliculares. Esta enzima participa nos processos de oxidação do iodo (conversão de iodeto em iodo), na sua organificação (ligação covalente do iodo aos resíduos tirosil da tiroglobulina) e na fusão das iodotirosinas dando origem às iodotironinas84. A primeira mutação foi descrita ainda nos anos 9085. Ao longo das últimas 2 décadas muitas outras mutações têm sido detetadas em doentes com HC, o que faz deste gene o principal candidato nos casos de dishormonogénese com défice de organificação de iodo e níveis elevados de TG circulante86–88. Tal como nos anteriores genes, a manifestação da doença requer a inativação de ambos os alelos. Em circunstâncias particulares de HC foi encontrada uma mutação de apenas um alelo, especulando‐se que possa existir uma mutação intrónica do alelo contralateral89–91. Não está, contudo, excluída a manifestação da doença em casos de heterozigotia, quer sob a forma efetiva quer aumentado a suscetibilidade para outros fatores, incluindo outras mutações genéticas83.
As NADPH oxidases tiroideias (THIOX1 e 2) são enzimas que participam na formação do peróxido de hidrogénio, imprescindível aos processos de oxi‐redução catalisados pela TPO92. Os 2 genes até agora descritos DUOX1 e 2 (dual oxidases) estão separados por um segmento de DNA com 16Kb. A DUOX1 e 2 partilham cerca de 83% dos aminoácidos, no entanto, a DUOX1 parece ter um papel menor na geração de peróxido de hidrogénio na glândula tiroideia. A DUOX2 é uma glicoproteína composta por 1.548 aminoácidos com múltiplos domínios intramembranares na região apical da célula folicular93. Ao contrário da maioria dos casos de dishormonogénese, as mutações heterozigóticas da DUOX2 podem ser suficientes para a manifestação da doença, habitualmente sob a forma de hipotiroidismo transitório neonatal, quando as necessidades de hormonas tiroideias são relativamente elevadas94–96. A reavaliação posterior destes doentes, após suspensão da levotiroxina tem revelado uma função tiroideia consistentemente normal, exceto em situações como a gravidez em que a necessidade de hormonas tiroideias está de novo aumentada. Em circunstâncias de inativação bialélica do gene DUOX2 tem sido demonstrada uma capacidade residual de produção de hormonas tiroideias ativas, pensa‐se que por ação da DUOX1. Admite‐se ainda que o iodo possa exercer uma ação moduladora sobre estes genes, pelo que a sua expressão pode ser influenciada por fatores ambientais como a disponibilidade em iodo97.
No processo de geração do peróxido de hidrogénio participam ainda 2 cofatores designados por fatores de maturação e que são codificados pelos genes denominados DUOXA1 e 2 (dual oxidase maturation factors)98. Estes genes ocupam uma posição intermédia entre os genes DUOX 1 e 2 e estão organizados em unidades funcionais com as respetivas oxidases95. Codificam uma proteína que intervém no processo de maturação e transição da DUOX1 e 2 do retículo endoplasmático para o complexo Golgi, bem como na sua posterior translocação para a membrana apical. Ao contrário do gene da DUOX 2, a manifestação clínica da doença sob a forma de HC ocorre apenas em situações de inativação bialélica do gene DUOXA299.
Através de um processo de pinocitose, a triiodotironina e a tiroxina ainda acopladas à TG penetram no citoplasma da célula folicular onde se fundem com os lisossomas. São as enzimas lisossomais que libertam as hormonas tiroideias da tiroglobulina, sendo de seguida exportadas para a corrente sanguínea através da membrana basal. O iodo remanescente que permanece ligado à TG sob a forma de mono e diiodotirosinas é então reciclado pela ação da enzima IYD (dehalogenase) localizada na membrana apical da célula folicular, bem como na membrana lisossomal100. O défice desta enzima origina uma excreção urinária aumentada de iodo sob a forma de mono e diiotirosinas com a consequente depleção de iodo, que se pode manifestar ainda durante os primeiros anos de vida sob forma de hipotiroidismo e bócio101. Caracteristicamente estes doentes apresentam uma fixação rápida e intensa do iodo radioativo nos cintigramas precoces da tiroideia, mas com um declínio também acelerado, traduzindo um «turnover» aumentado do iodo. As mutações deste gene até agora descritas transmitem‐se de forma autossómica recessiva.
Tratamento do hipotiroidismo congénitoAbordagem inicialApós a confirmação do HC, o recém‐nascido deve iniciar de imediato a terapêutica com levotiroxina (tabela 3). O objetivo é normalizar a T4 em 2 semanas e o TSH em um mês102. A dose inicial de levotiroxina recomendada é de 10‐15μg/kg dependendo da gravidade do HC. A administração «per os» de 50μg/dia de levotiroxina é suficiente para normalizar a T4 livre em 3 dias e o TSH em cerca de 2 semanas. A normalização do TSH pode eventualmente ser mais demorada, atendendo a que existe uma resistência relativa da hipófise durante os primeiros meses de vida como consequência do hipotiroidismo durante a gravidez. O comprimido de levotiroxina deve ser triturado e dissolvido em alguns ml de leite ou água. Deve evitar‐se a administração simultânea de soja, fibras ou de fármacos como o cálcio e o ferro. A amamentação deve prosseguir normalmente. A T4 livre deve ser mantida no limite superior do normal e o TSH no limite inferior do normal, durante os primeiros 3 anos de tratamento. É fundamental monitorizar a função tiroideia, atendendo a que crianças com níveis de T4 l abaixo do limite inferior normal têm um desenvolvimento psicomotor subótimo. Níveis de T4 acima destes limites estão associados a alterações de comportamento e a casos de craniosinostose. Na maioria dos casos a elevação persistente do TSH é devida a uma fraca adesão á terapêutica. De salientar que 4 ou mais determinações de TSH com valores superiores a 6mU/L durante os primeiros 6 meses de vida, estão associados a um atraso escolar relevante. Neste sentido, a terapêutica com levotiroxina no recém‐nascido deve ser ajustada de uma forma rigorosa e as eventuais causas de má absorção intestinal ou de fraca adesão ao tratamento devem ser discutidas com os progenitores da criança. Fármacos como o ferro ou cálcio, alimentos ricos em fibras e a soja, são as causas mais frequentes de uma deficiente absorção intestinal da levotiroxina. Por sua vez, os anticonvulsivantes aceleram a degradação da levotiroxina. Por este motivo, nas crianças com epilepsia a dose de levotiroxina deve ser mais elevada. Em circunstâncias raras como nos hemangiomas de grandes dimensões em que atividade de desiodase da T4 em T3 está aumentada, os valores de T4 podem manter‐se baixos, mas com uma T3 livre elevada. Só nestas circunstâncias está indicada a determinação simultânea do T4 e T3 livres na avaliação da terapêutica com levotiroxina no recém‐nascido.
Abordagem do hipotiroidismo congénito
| ABORDAGEM INICIAL IMEDIAMENTE APÓS A REFERENCIAÇÃO |
| História clínica e exame objetivo detalhados |
| Colheita de sangue venoso para determinação de TSH e T4 livre |
| Ecografia |
| EM CASO DE CONFIRMAÇÂO DO HIPOTIROIDISMO CONGÉNITO |
| Levotiroxina 10‐15μg/kg de peso per os, uma vez por dia |
| MONITORIZAÇÃO |
| Reavaliar TSH e T4 livre |
| ‐ Às 2 e 4 semanas após o início do tratamento |
| ‐ De 2 em 2 meses nos primeiros 6 meses |
| ‐ De 3 em 3 meses até aos 3 anos |
| ‐ Semestral/Anual até ao final do crescimento |
| OBJETIVO DA TERAPÊUTICA |
| Normalizar o TSH e manter o T4 livre na metade superior do intervalo de referência |
O desempenho intelectual e o desenvolvimento físico nos doentes com HC adequadamente tratados são semelhantes ao das outras crianças e adultos103. Todos os esforços devem ser feitos no sentido de que este objetivo seja concretizado. A avaliação clínica dos recém‐nascidos com HC e a monitorização da função tiroideia deve ser feita às 2 e 4 semanas após o início do tratamento, de 2 em 2 meses até aos 6 meses de idade e a cada 3 meses durante os primeiros 3 anos de vida, altura em que as avaliações periódicas podem ser espaçadas para intervalos de 6 meses até ao final do crescimento. Os intervalos recomendados para a avaliação da função tiroideia estão resumidos no tabela 3. Como referido anteriormente, o HC está associado a uma percentagem muito mais elevada de malformações congénitas do que a população em geral, isto é, cerca de 3 vezes mais. Entre as mais frequentes estão as malformações cardiovasculares incluindo a estenose pulmonar e os defeitos do septo auricular e ventricular. Qualquer suspeita clínica deve ser investigada com recurso aos exames complementares adequados. O estrabismo bem como a surdez neurossensorial são também mais comuns nos doentes com HC e devem ser rastreados logo que possível. As malformações génito‐urinárias merecem também particular atenção no contexto dos HC provocados por mutações do gene PAX8.
Avaliação do hipotiroidismo permanenteA presença de uma tiroideia ectópica num recém‐nascido garante‐nos que o hipotiroidismo é permanente. Quando a causa de HC não é evidente após a realização dos exames morfológicos e os valores de TSH se mantenham normais ao longo de todo o período neonatal, a administração de levotiroxina deve ser suspensa durante um mês, após os 3 anos de idade. Se após um mês sem terapêutica o valor da T4 livre descer para valores abaixo do normal em simultâneo com uma subida do TSH confirma‐se a presença de hipotiroidismo permanente e o tratamento com levotiroxina é reiniciado. Se pelo contrário a TSH e a T4 livre se mantiverem normais, o diagnóstico de hipotiroidismo transitório é então o mais provável e a terapêutica com levotiroxina deve ser suspensa. Nesta última situação é da maior importância que a criança mantenha um seguimento regular e que os testes de função tiroideia sejam repetidos periodicamente. A terapêutica deve ser reinstituída caso seja detetada uma subida do TSH, mesmo que ligeira. Nos casos em que após a suspensão da terapêutica os valores de T4 livre e TSH sejam inconclusivos, a opção é reiniciar o tratamento e reavaliar periodicamente.
ConclusõesCom o advento do rastreio neonatal, o HC é hoje uma doença facilmente diagnosticada e com uma terapêutica eficaz capaz de prevenir as sequelas major da doença.
Nunca é de mais realçar que algumas formas HC podem escapar ao diagnóstico precoce. É o caso dos HC de causa central que cursam com hipotiroxinemia, mas com valores TSH baixos ou indoseáveis. Esta situação é particularmente relevante nos recém‐nascidos prematuros, atendendo a que nestas circunstâncias a subida do TSH pode demorar algumas semanas ou até meses. É justamente por isso que tem sido equacionada a necessidade de realização de um segundo teste de rastreio nos recém‐nascidos prematuros não referenciados, entre 2‐4 semanas após o primeiro. Em conclusão, o facto de um recém‐nascido não ter sido referenciado não é uma condição «sine qua non» para isentá‐lo de patologia tiroideia, pelo que os clínicos devem manter‐se atentos para a possibilidade da ocorrência de HC ou de outras formas de hipotiroidismo com início na infância, não necessariamente de origem congénita, em particular na avaliação de situações de má progressão estaturo‐ponderal e de atraso no desenvolvimento psicomotor.
Uma vez diagnosticado o HC, a terapêutica com levotiroxina, deve ser iniciada o mais precocemente possível, numa dose de 0,10‐0,15mcg/kg/dia, capaz de normalizar a T4 livre e o TSH em 2‐4 semanas. A monitorização subsequente da função tiroideia deve ser enquadrada num plano de «follow‐up» destes doentes com consultas regulares programadas a cada 3 meses, até pelo menos aos 3 anos de idade. Finalmente, uma chamada de atenção para a necessidade do despiste de malformações congénitas nos casos de HC, uma vez que a sua incidência nestes doentes é cerca de 3 vezes superior à da população em geral.
Conflito de interessesOs autores declaram não haver conflito de interesses.
