


A síndrome de Cushing resulta de exposição crónica a níveis excessivos de glicocorticoides. A sua investigação e abordagem continua a ser um desafio diagnóstico, uma vez que, por um lado, apresenta um espectro clínico abrangente que inclui sintomas e sinais pouco específicos como o excesso ponderal, depressão, HTA, alteração do metabolismo dos hidratos de carbono, o que dificulta o diagnóstico em fase precoce da doença, por outro lado, a síndrome de Cushing de causa endógena associa‐se a uma grande variedade de patologias com baixa prevalência. Apesar de cada vez mais clínicos considerarem este diagnóstico, a falta de articulação entre os profissionais e uma visão pouco global do paciente conduz em alguns casos a diagnóstico tardio, com impacto sobre a morbilidade e mortalidade. Na ausência de tratamento a taxa de mortalidade aumenta 50% em 5 anos1.
Neste artigo apresentamos o caso de uma doente com quadro exuberante de síndrome de Cushing, com diagnóstico tardio e consequente atrofia do córtex da suprarrenal contralateral com necessidade prolongada de terapêutica de substituição.
A propósito deste caso clínico revemos os aspetos mais importantes de rastreio, diagnóstico e tratamento de síndrome de Cushing. Adicionalmente revemos os aspetos relacionados com prognóstico.
Cushing's Syndrome results from chronic exposure to excessive levels of glucocorticoids. It's investigation and management remains an on‐going challenge in clinical endocrinology. It is a rare condition with a broad clinical spectrum, including non‐specific symptoms such as obesity, depression and muscle weakness with high prevalence in general population, what hinders the diagnosis. Despite the clinicians considering this diagnosis more often, the lack of articulation between professionals and poor global vision about the patient leads in some cases to a late diagnosis, with impact on morbidity and Mortality. In Cushing Syndrome the mortality is increased 50% in 5 years1.
In this paper we present a case of exuberant Cushing's syndrome, with late diagnosis, and atrophy of the adrenal cortex with consequent need of replacement therapy after treatment.
We review the most important aspects of screening, diagnosis and treatment of Cushing's syndrome and aspects related with prognosis.
A síndrome de Cushing resulta da exposição crónica a níveis excessivos de glicocorticoides. A síndrome de Cushing endógena é rara, com uma incidência anual de 0,1‐1,0 novos casos por 100.000 indivíduos1.
Os sintomas e sinais que a caracterizam dependem do grau de elevação do cortisol e tempo de exposição. Em fase precoce o espectro clínico inclui sintomas e sinais pouco específicos e muito prevalentes na população geral, como o excesso ponderal, hipertensão arterial (HTA), alteração do metabolismo dos hidratos de carbono, depressão, de que resultam 2 implicações: necessidade frequente de investigação e dificuldade de diagnóstico em fase precoce da doença.
O aparecimento destas características em simultâneo e, posteriormente, o desenvolvimento de sinais mais específicos (em particular a distribuição do tecido adiposo central, miopatia proximal, fragilidade cutânea, alterações menstruais/fertilidade, perda acelerada de densidade mineral óssea e fraturas de fragilidade em indivíduos sem fatores de risco conhecidos) deverão fazer suspeitar o diagnóstico.
A síndrome de Cushing pode ser causada por uma grande variedade de patologias raras, que podem ser agrupadas em síndrome de Cushing ACTH dependente e hormona adrenocorticotrófica (ACTH) independente.
A síndrome de Cushing ACTH independente é a menos frequente, responsável por apenas 15% dos casos2. Neste grupo inclui‐se o adenoma e carcinoma do córtex da suprarrenal e as hiperplasias micronodulares pigmentadas (esporádica ou integrada no complexo de Carney) e macronodulares (associada frequentemente a síndrome de Cushing cíclica)3,4.
A falta de articulação entre os profissionais e uma visão pouco global do paciente conduz em alguns casos a diagnóstico tardio, com impacto sobre a morbilidade.
Apresentamos o caso de uma doente com quadro exuberante de síndrome de Cushing, associada a adenoma do córtex da suprarrenal, em que o diagnóstico foi tardio, condicionando a atrofia do córtex da suprarrenal contralateral e necessidade de terapêutica de substituição.
A propósito deste caso clínico revemos os aspetos mais importantes de rastreio, diagnóstico, tratamento e prognóstico da síndrome de Cushing.
Relato do caso clínicoDoente de 29 anos, sexo feminino, assintomática até aos 25 anos, altura em que iniciou oligomenorreia, sem outros sintomas associados.
Por indicação do ginecologista assistente iniciou um anticoncetivo oral combinado (ACO) com regularização dos ciclos menstruais. Na altura fez avaliação laboratorial e ecográfica que «não terá mostrado alterações» (sic).
No ano seguinte suspendeu o ACO com o objetivo de engravidar. Após cerca de um ano sem sucesso recorreu a uma clínica de fertilização e engravidou em resultado de tratamento hormonal.
Nessa altura, apresentava um aumento ponderal significativo, com distribuição adiposa central, pletora facial, que não valorizou. No mesmo ano diagnosticou‐se HTA, iniciou combinação de 2 anti‐hipertensores com controlo tensional adequado. Não foram investigadas causas secundárias.
Durante a gestação notou o aparecimento de extensas estrias abdominais violáceas, equimoses fáceis nas mãos, braços e pernas, fraqueza muscular proximal intensa, que a limitava nas atividades quotidianas, em particular em tarefas que implicavam subir escadas e utilizar os membros superiores. No final do segundo trimestre de gravidez diagnosticou‐se diabetes mellitus gestacional, que controlou com a dieta. O parto foi de termo, por cesariana, o recém‐nascido pesava 3,1kg, não apresentava malformações. Teve um apgar 9/10. Amamentou 4 meses.
No pós‐parto apresentou infeção do local de sutura e iniciou quadro de lombociatalgia direita intensa, com limitação da marcha (necessidade de auxílio com canadianas), refratária à terapêutica. Alguns meses depois foi observada por reumatologia e ortopedia. Os estudos radiológico e densitométrico identificaram: diminuição da massa óssea (Zscore coluna Lombar – 2,2DP), fraturas compressivas em diversas vértebras dorsais e lombares (D10, 11, 12 e L4 e L5), fratura da bacia em 3 locais diferentes (ramos ílio‐púbicos esquerdo e direito e ramo isquiopúbico à esquerda), pelo que iniciou alendronato e suplementação de cálcio e vitamina D.
Referiu ainda início de queixas depressivas e cefaleia frontal intensa, tipo opressivo, com pouca resposta à medicação analgésica. O quadro foi interpretado como depressão pós‐parto, tendo iniciado venlafaxina 75mg/dia e clorazepato dipotássico 10mg ao deitar, sem melhoria significativa.
A doente não apresentou história familiar ou pessoal de risco para patologia endocrinológica ou neoplásica.
Posteriormente, foi observada em consulta de endocrinologia, onde acorreu com o objetivo de normalizar o peso. Aí identificou‐se hipercortisolismo (cortisol urinário de 24h de 693,7ug/dL (<286) e foi proposto internamento eletivo no serviço de endocrinologia para investigação de síndrome de Cushing (suspendeu anticoncetivos orais nas 4 semanas anteriores).
À data de admissão do exame objetivo salientava‐se:
- •
obesidade (IMC 32) com tecido adiposo de distribuição central (face em meia lua, depósitos de gordura supraclavicular, aumento do perímetro abdominal – 114cm [<80]);
- •
hipertricose muito ligeira na face, sem acne. Sem áreas de alopecia;
- •
pele fina e equimoses extensas na região pré‐tibial. Estrias abdominais longas com cerca de 3cm de largura, violáceas. Cicatriz abdominal vertical mediana, rosada;
- •
massa muscular dos membros superiores e inferiores com atrofia simétrica e fraqueza muscular proximal;
- •
cifoescoliose;
- •
normotensão sob anti‐hipertensor;
- •
eutímica, com labilidade emocional.
Estabeleceu‐se hipercortisolismo endógeno‐autónomo pela seguinte avaliação laboratorial:
- •
dois doseamentos de cortisol urinário, o mais elevado de 793ug/24h (<286);
- •
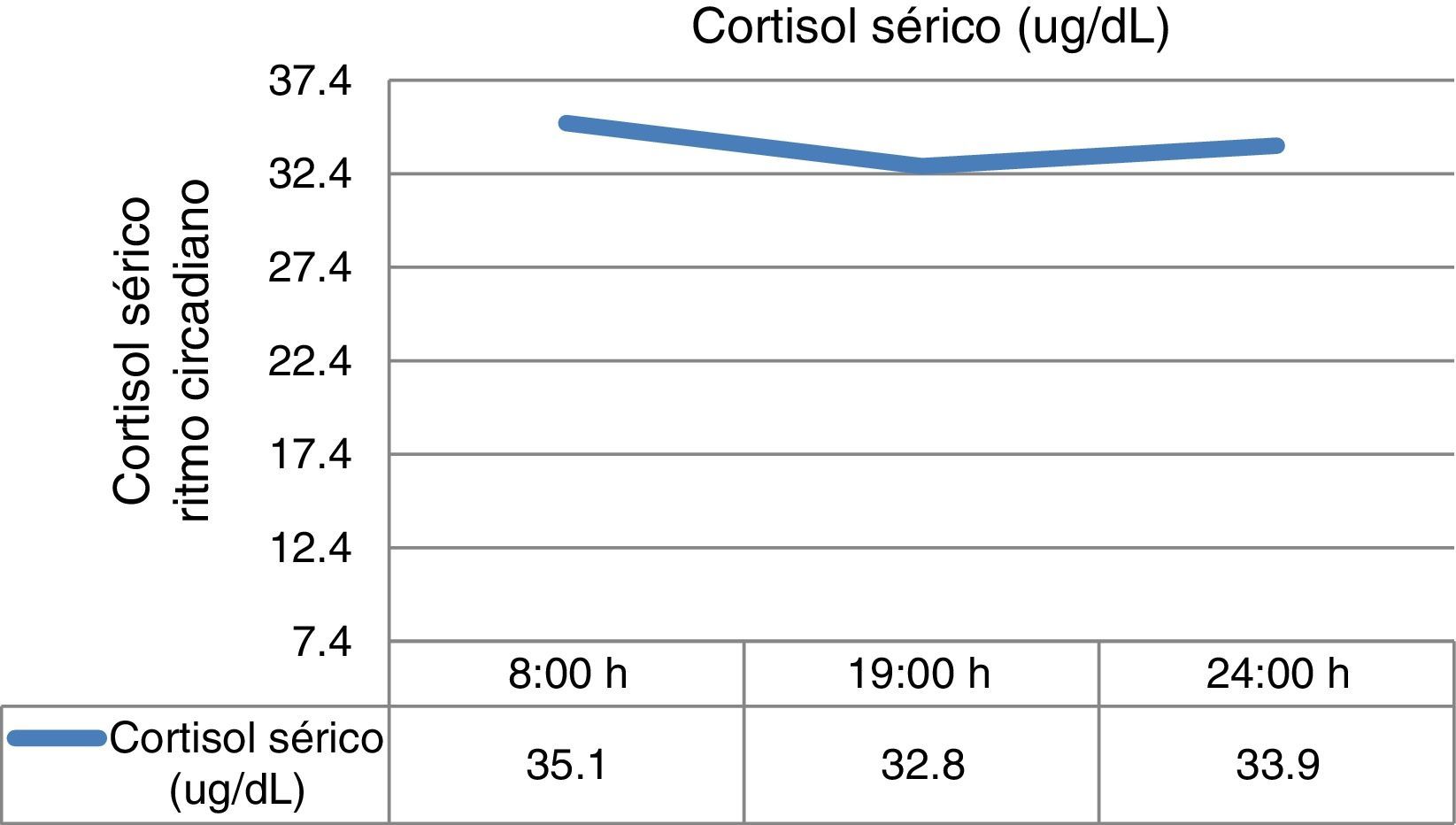
cortisol sérico entre as 23‐24:00h: 33,9ug/dL (<7,5ug/dL);
- •
doseamentos de cortisol sérico seriados revelando ausência de ritmo circadiano (fig. 1).

Após administração de dexametasona não houve supressão do eixo hipotálamo‐hipófise‐corticoadrenal:
após 1mg dexametasona: cortisol sérico 8‐9h da manhã, em jejum – 34,0ug/dL (<1,8ug/dL);
após administração de 0,5mg de dexametasona 6‐6h durante 48h: cortisol sérico 8‐9h, jejum – 31,1ug/dL (<1,8).
Excluiu‐se patologia que pudesse interferir com o metabolismo dos glicocorticoides, como insuficiência renal e hepática.
Dois doseamentos de ACTH sérico revelaram valores inferiores a 5pg/mL, o que nos permitiu estabelecer o diagnóstico de síndrome de Cushing ACTH‐independente, de provável causa adrenal.
Para localização da doença foi pedida uma tomografia computorizada abdominal que revelou na «suprarrenal direita nódulo de 31x22mm, com contornos regulares, elevada densidade (30‐35HU), que realça significativamente após contraste e precocemente esboça lavagem. Suprarrenal esquerda sem nódulos».
A densitometria óssea e avaliação laboratorial do metabolismo ósseo confirmaram o diagnóstico de osteoporose.
A doente foi proposta para adrenalectomia. O exame anatomopatológico da peça operatória revelou «peça de suprarrenalectomia, com nódulo de 2,8cm de maior eixo, bem circunscrito, que histologicamente corresponde a adenoma do córtex da suprarrenal».
No primeiro dia pós‐operatório o cortisol 8‐9h manhã foi de 1,0ug/dL (4,3‐23).
A doente teve alta, sob terapêutica de substituição, com 15mg/dia de hidrocortisona e foi observada em consulta endocrinologia com a seguinte periocidade: 10 e 18 dias após a cirurgia, onde não se observaram sinais de excesso de cortisol, e ainda aos 5, 9, e 16 meses após cirurgia, tendo feito prova de Synacthen com respostas inadequadas.
A última prova, 19 meses após cirurgia, revelou reserva adrenal insuficiente (ACTH basal 82,0pg/mL, cortisol basal 13,2ug/dL, cortisol 30min 14,8ug/dL, 60min 15,9ug/dL), pelo que manteve terapêutica de substituição.
Dezasseis meses após o tratamento apresentava perda de 14kg e ausência de pletora facial. Mantinha estrias claras, excesso e laxidão cutânea a nível axilar, braquial e abdominal.
Verificou‐se ainda normalização do perfil tensional e lipídico sem medicação, sob dieta hipossalina‐hipocalórica.
Regularizou os ciclos menstruais.
Inicialmente foi medicada com alendronato, com variação não significativa da Densidade Mineral Óssea (DMO), pelo que se optou por realizar ácido zoledrónico em associação a suplemento de cálcio e vitamina D.
Realiza exercício de carga pelo menos uma hora, 3 vezes por semana (caminhada) e fisioterapia.
Mantém lombociatalgia sem indicação para cirurgia, segundo neurocirurgia.
Mantém labilidade emocional, mas com melhoria significativa do quadro depressivo inicial. Recusou psicofármacos e psicoterapia.
ComentárioApresentamos uma doente de 29 anos, enviada à consulta de endocrinologia com quadro de cerca de 3 anos de evolução, instalação insidiosa e agravamento progressivo. O quadro foi exacerbado no período gestacional.
No pós‐parto imediato apresentou infeção da sutura cirúrgica. Adicionalmente, a doente apresentava hipertensão diagnosticada aos 26 anos e diminuição significativa da DMO em comparação com indivíduos da mesma faixa etária, com múltiplas fraturas, o que sugeria causa secundária para ambas.
A doente apresentava uma síndrome depressiva, com início concomitante ao restante quadro clínico e com pouca resposta a psicofármacos. Negava hábitos alcoólicos ou consumo de fármacos com atividade glicocorticoides.
O início abrupto de hipertensão grave (inferida pela necessidade da combinação de 2 anti‐hipertensores) em idade inferior a 35 anos sugeria uma HTA secundária.
A DMO diminuída para o grupo etário (Z score ≤–2,2DP), com fraturas vertebrais assintomáticas e da bacia durante o parto permitiu‐nos estabelecer o diagnóstico de osteoporose secundária.
Relativamente à suspeita de síndrome de Cushing, os dados que sugeriam o diagnóstico eram:
- •
sexo (a síndrome de Cushing é 8 vezes mais frequente em mulheres);
- •
idade (pico 20‐40 anos);
- •
quadro clínico muito sugestivo de síndrome de Cushing, incluindo algumas das características consideradas mais específicas, miopatia proximal, estrias violáceas e equimoses fáceis;
- •
a infeção da sutura cirúrgica, a sugerir imunodepressão.
A causa mais frequente de síndrome de Cushing é a administração exógena de corticoides, que foi excluída nesta doente.
Na investigação, o primeiro passo foi confirmar a existência de hipercortisolismo endógeno autónomo, o que nos permitiu distinguir síndrome de Cushing endógena, de patologia associada a hipercortisolismo não autónomo (pseudo Cushing).
As 2 principais causas de pseudo Cushing são a síndrome depressiva e o alcoolismo5. A doente apresentava uma síndrome depressiva, sendo que 80% dos indivíduos com depressão major apresentam algum grau de hipercortisolismo, no entanto, o cortisol urinário de 24h raramente ultrapassa os 300ug/24h, como nesta doente5. O facto de a depressão ter início concomitante ao restante quadro clínico e ser refratária aos psicofármacos, favorecia a suspeita de síndrome de Cushing1,4.
Neste caso, através de dados laboratoriais, confirmou‐se o hipercortisolismo endógeno‐autónomo.
No entanto, quando os resultados não são inequívocos, a prova combinada de dexametasona e Hormona Libertadora de Corticotrofina (CRH) permite distinguir os indivíduos saudáveis ou com pseudo Cushing (15 minutos após administração de CRH, mantém a supressão – cortisol <1,4ug/dL) daqueles com verdadeira síndrome de Cushing (elevação do cortisol >1,4ug/dL), com cerca de 100% de sensibilidade e especificidade5.
Na investigação da etiologia da síndrome de Cushing as causas a considerar podem ser divididas em 2 grandes grupos, dependendo da sua relação com a ACTH.
A síndrome de Cushing ACTH‐dependente, é mais comum, responsável por cerca de 85% dos casos de hipercortisolismo endógeno. A síndrome de Cushing ACTH‐independente, como no caso clínico apresentado, é responsável por cerca de 15% dos casos de hipercortisolismo endógeno2.
A diferenciação entre estes grupos faz‐se através de 2 doseamentos de ACTH sérico. Doseamentos inferiores a 5pg/mL estabelecem um mecanismo ACTH‐independente5.
São causas mais raras de síndrome de Cushing ACTH‐independente: hiperplasia adrenal macronodular e hiperplasia adrenal micronodular pigmentada (esporádica ou em contexto de complexo de Carney)3,4. O tumor da suprarrenal é a causa mais frequente. Nestes casos, é importante diferenciar o adenoma (77%) do carcinoma (33%)1,4.
Os seguintes aspetos sugeriam benignidade:
- •
quadro de instalação insidiosa, sem hiperandrogenismo (ausência de sinais de virilização e níveis de SDHEA, androstenediona e testosterona total dentro de valores fisiológicos).
- •
no exame de imagem, diâmetro tumoral inferior a 4cm, contornos regulares, homogéneo, captação rápida de contraste com eliminação também rápida6. A elevada atenuação (30‐35HU) é uma característica a favor de malignidade, mas considerando os aspetos referidos acima esta seria uma hipótese muito pouco provável.
Nestes caso, porque a doente não apresenta história pessoal ou familiar sugestiva e pelas razões expostas a seguir, a hipótese de síndrome hereditária‐poliglandular foi considerada muito pouco provável.
Os adenomas da suprarrenal podem surgir integrados em 3 síndromes hereditárias: síndrome MEN1, polipose adenomatosa familiar (clássica ou associada a síndrome de Gardner), na síndrome de McCune–Albright4.
Nos casos de adenoma do córtex adrenal, a ressecção cirúrgica completa apresenta uma taxa de cura de 100%, quando o cirurgião é experiente (mínimo de 20 adrenalectomias/ano)1. A recidiva é muito rara e quando ocorre sugere carcinoma adrenal, pelo que a revisão de lâminas deverá ser pedida.
Neste caso, o cortisol do primeiro dia pós‐operatório de 1ug/dL e a necessidade de terapêutica de substituição são fatores de bom prognóstico4.
Em conclusão:
apresentamos um caso de adenoma da suprarrenal produtor de cortisol, com diagnóstico tardio, apesar de quadro clínico exuberante.
A exposição prolongada a níveis de cortisol elevados condicionou:
- •
na fase pré‐tratamento:
- ∘
oligomenorreia e subfertilidade, por disfunção do eixo hipotálamo‐hipófise‐adrenal‐gónadas;
- ∘
aumento do risco cardiovascular associado a obesidade e HTA e dislipidemia de difícil controlo com terapêutica médica;
- ∘
síndrome depressiva, resistente à terapêutica;
- ∘
osteoporose fraturária;
- ∘
imunossupressão.
- ∘
- •
Na fase pós‐tratamento:
- ∘
necessidade de terapêutica substitutiva por inibição prolongada do eixo hipotálamo‐hipófise e atrofia da córtex adrenal contralateral.
- ∘
Com a publicação deste caso pretendemos alertar para a existência desta síndrome, divulgar os sintomas e sinais mais específicos que deverão sugerir o diagnóstico e ainda reforçar a importância de realizar testes de rastreio perante suspeita clínica adequada. Desta forma, as síndromes de Cushing poderão ser referenciadas a consulta especializada precocemente, o que terá impacto positivo sobre a morbilidade e mortalidade dos pacientes.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dadosOs autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes.
Direito à privacidade e consentimento escritoOs autores declaram que não aparecem dados de pacientes neste artigo.
Conflito de interessesOs autores declaram não haver conflito de interesses.
