




Los tumores malignos de la vaina del nervio periférico representan del 5 al 10% del total de tumores malignos de tejidos blandos y frecuentemente se asocian con la neurofibromatosis tipoI. Su presentación en la glándula mamaria es poco frecuente, y el diagnóstico es muy difícil por su rareza y por la ausencia de características clínicas y/o radiológicas específicas, siendo preciso una adecuada interpretación del estudio inmunohistoquímico y su correlación con los hallazgos morfológicos. Presentamos un caso clínico y su revisión bibliográfica.
Malignant peripheral nerve sheath tumours represent 5-10% of all malignant soft tissue tumours and are frequently associated with neurofibromatosis typeI. Their presentation in the mammary gland is rare. Because of this rarity, as well as the absence of specific clinical and/or radiological features, their diagnosis very difficult. It is therefore essential to correctly interpret immunohistochemical analysis and its correlation with morphological findings. We present a clinical case and literature review.
Los tumores de la vaina del nervio periférico son lesiones que derivan de las células de Schwann o de las células pluripotenciales de la cresta neural. Comprenden tumores benignos como el schwannoma y el neurofibroma, y malignos como el tumor maligno de la vaina del nervio periférico (TMVNP)1.
Los TMVNP representan del 5 al 10%2 del total de tumores malignos de tejidos blandos. La mitad de los casos se asocian a una neurofibromatosis tipoI3,4, el 11% con haber recibido tratamiento radiante previo4,5 y el 40% restante suceden de forma esporádica. Se manifiestan con mayor frecuencia en tronco, extremidades, cabeza y cuello; la presentación en la mama es extremadamente rara.
Describimos a continuación un caso de TMVNP primario de mama diagnosticado en nuestro centro y su revisión bibliográfica.
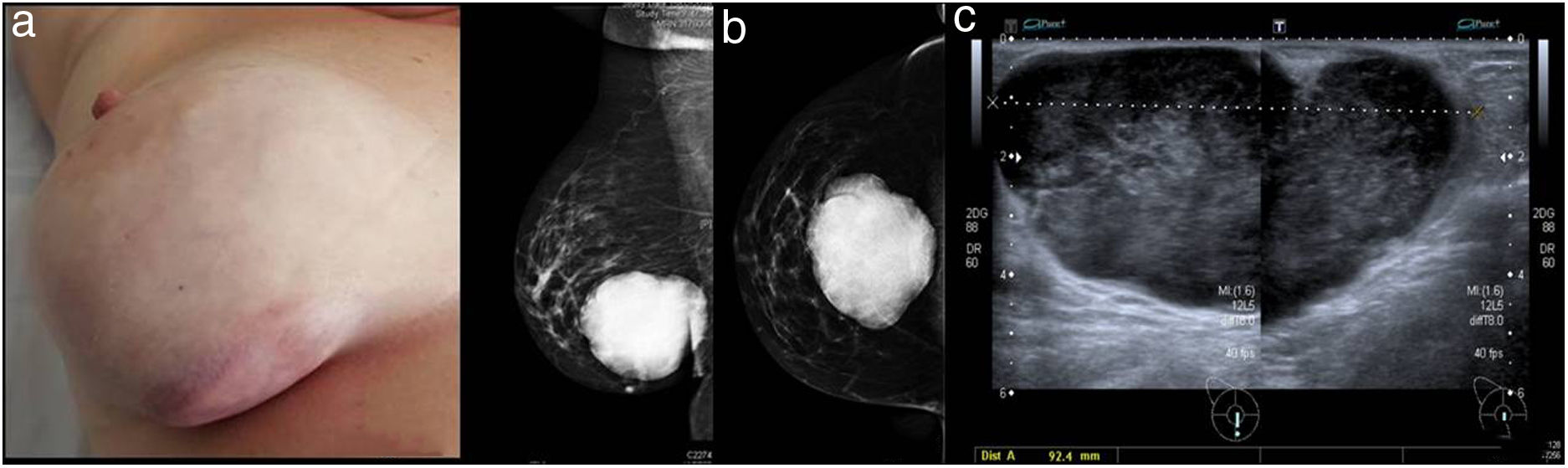
Caso clínicoMujer de 48 años, sin antecedentes familiares ni personales de interés, que consulta por presentar una lesión nodular palpable en mama derecha no dolorosa y de rápido crecimiento en los últimos 2meses. En la exploración física se objetivó una asimetría mamaria a expensas de un nódulo duro, irregular, de 10×10cm presente en los cuadrantes inferiores de la mama derecha próximo a piel, sin adenopatías regionales patológicas asociadas (fig. 1a). El estudio radiológico mamario informó de la presencia de una lesión nodular irregular de margen circunscrito de 92mm en el cuadrante inferior de la mama derecha, BIRADS 5 (fig. 1b,c), y el estudio de extensión fue negativo para afectación metastásica a distancia. En el estudio histológico, tras la biopsia con aguja gruesa se observó una proliferación estromal fusocelular con atipia leve y bajo-moderado índice proliferativo, sin identificar componente epitelial. La celularidad tumoral fue negativa para CKAE1/AE3, CD31, CD34, receptores de estrógenos y progesterona, actina, desmina, miogenina, CK5/6, GFPA, neurofilamentos y p63, y positiva para S-100. Los hallazgos se consideraron compatibles con el diagnóstico de schwannoma, por lo que se practicó la exéresis completa de la lesión.
exploración física; b)mamografía (proyección MLO y CC) de mama derecha: nódulo irregular, de margen circunscrito e hiperdenso en los cuadrantes inferiores de la mama derecha. BIRADS 5; c)ecografía de mama derecha: nódulo de 92mm ovalado, con margen circunscrito y orientación paralela a la piel, con refuerzo acústico posterior y estudio axilar sin hallazgos. BIRADS 5.")
Exploración clínica y radiológica: a)exploración física; b)mamografía (proyección MLO y CC) de mama derecha: nódulo irregular, de margen circunscrito e hiperdenso en los cuadrantes inferiores de la mama derecha. BIRADS 5; c)ecografía de mama derecha: nódulo de 92mm ovalado, con margen circunscrito y orientación paralela a la piel, con refuerzo acústico posterior y estudio axilar sin hallazgos. BIRADS 5.
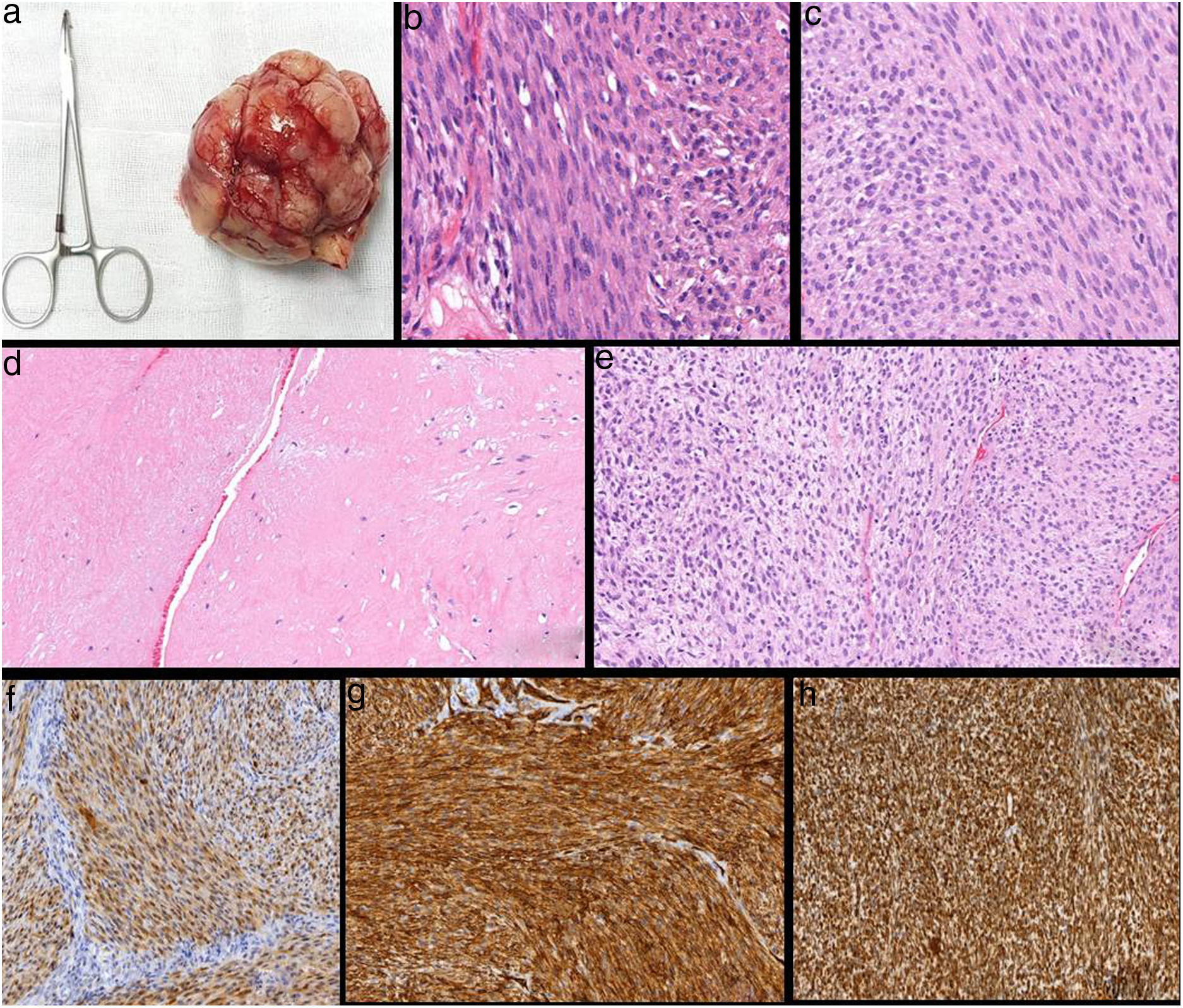
El estudio anatomopatológico definitivo de la pieza describió macroscópicamente una lesión nodular no encapsulada de 9×7,8×5cm y 192g de peso (fig. 2a), constituida microscópicamente por una proliferación fusocelular con atipia leve-moderada, núcleos de aspecto vesiculoso con nucléolos evidentes y presencia de áreas con abundantes mitosis (hasta 15/10CGA). La celularidad se disponía en forma de fascículos o haces entrecruzados, alternando áreas hipo e hipercelulares, presentando áreas de patrón estoriforme que formaban pseudoempalizadas perivasculares. El estudio inmunohistoquímico mostró expresión positiva para vimentina, CD34 y S-100, siendo negativa para CK AE1/AE3, p63, desmina, actina, HHF35, CD56, neurofilamentos, HMB45, CD99, factor VIII, BCL2 y EMA, y el Ki 67 fue del 20% (fig. 2b-h). Los hallazgos morfológicos e inmunohistoquímicos fueron compatibles con un TMVNP que contacta con los márgenes quirúrgicos, por lo que, tras comentar el caso clínico en el Comité Multidisciplinar de Tumores de Patología Mamaria, se indicó la realización de una mastectomía simple y radioterapia adyuvante. El control clínico y radiológico con TAC semestral no mostró recurrencias local ni a distancia tras un año de seguimiento.
. a)macroscópico: pieza quirúrgica de la exéresis de la lesión; b)tinción hematoxilina-eosina (HE) (20×); células fusiformes en empalizada. c)HE (20×); núcleos con aspecto vesiculoso con nucléolo evidente. d)HE (20×); áreas hipocelulares; e)HE (20×))áreas hipercelulares; f)IHQ positiva para S-100; g)IHQ positiva para CD34; h)IHQ positiva para vimentina.")
Estudio histopatológico-inmunohistoquímico (IHQ). a)macroscópico: pieza quirúrgica de la exéresis de la lesión; b)tinción hematoxilina-eosina (HE) (20×); células fusiformes en empalizada. c)HE (20×); núcleos con aspecto vesiculoso con nucléolo evidente. d)HE (20×); áreas hipocelulares; e)HE (20×))áreas hipercelulares; f)IHQ positiva para S-100; g)IHQ positiva para CD34; h)IHQ positiva para vimentina.
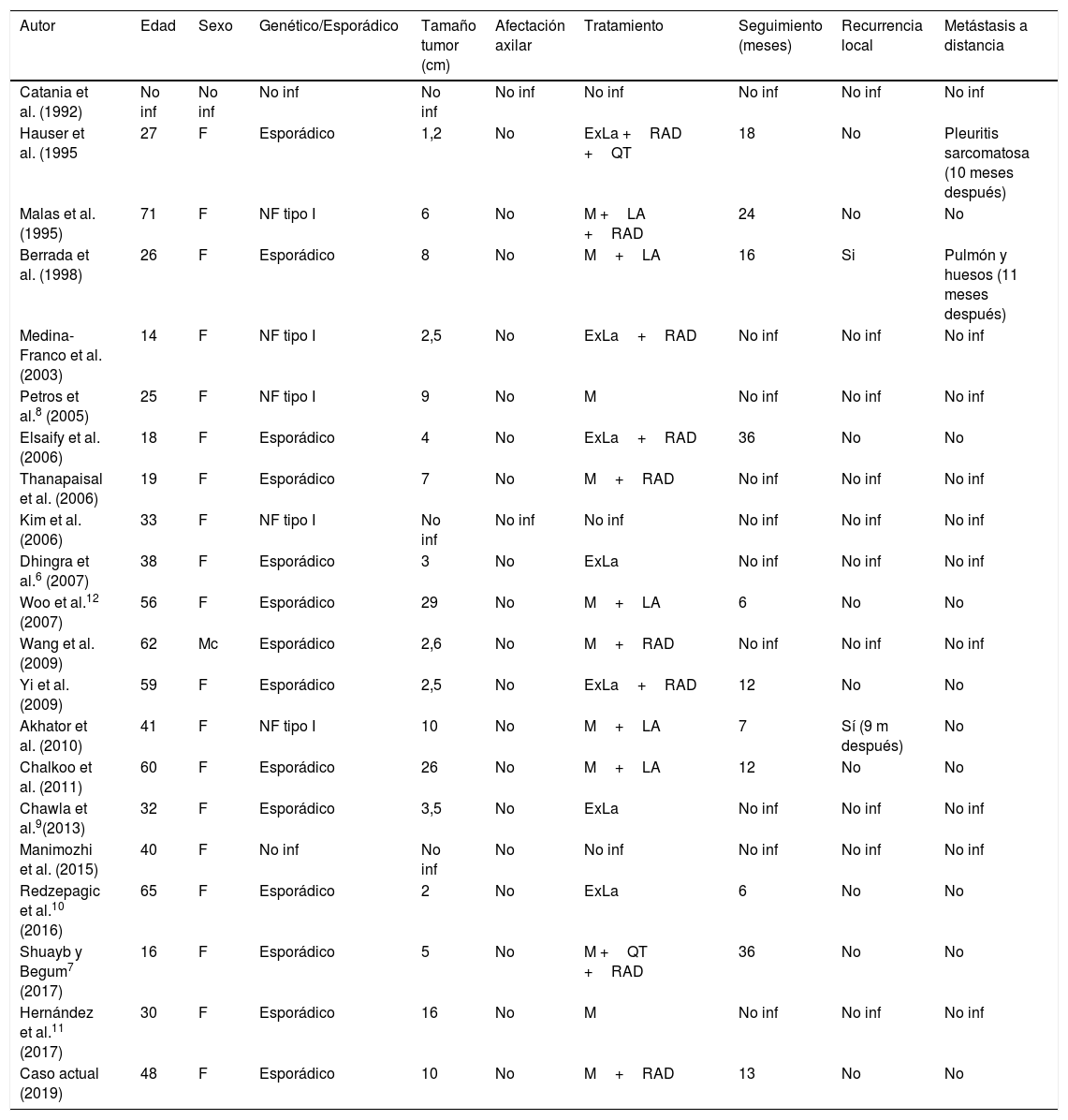
El término TMVNP sustituye términos antiguamente utilizados, como schwannoma maligno, neurilemmoma maligno y neurofibrosarcoma6. Su incidencia en la población general es del 0,001%5, y se pueden manifestar de forma aislada, asociados a neurofibromatosis tipoI o a tratamiento radiante previo4. La edad de presentación está descrita entre los 20 y los 50 años, aunque en los casos de pacientes con neurofibromatosis tipoI puede aparecen más precozmente5 y lo hace en forma de nódulo de partes blandas con un tamaño que suele ser >5cm5, normalmente en tronco, extremidades, cabeza y cuello, siendo la presentación en la mama extremadamente rara, con solo 20casos de TMVNP primario de mama descritos en la literatura (tabla 1)7-12.
Resultados clínicos de pacientes con TMVNP primario de mama en estudios previos
| Autor | Edad | Sexo | Genético/Esporádico | Tamaño tumor (cm) | Afectación axilar | Tratamiento | Seguimiento (meses) | Recurrencia local | Metástasis a distancia |
|---|---|---|---|---|---|---|---|---|---|
| Catania et al. (1992) | No inf | No inf | No inf | No inf | No inf | No inf | No inf | No inf | No inf |
| Hauser et al. (1995 | 27 | F | Esporádico | 1,2 | No | ExLa +RAD +QT | 18 | No | Pleuritis sarcomatosa (10 meses después) |
| Malas et al. (1995) | 71 | F | NF tipo I | 6 | No | M +LA +RAD | 24 | No | No |
| Berrada et al. (1998) | 26 | F | Esporádico | 8 | No | M+LA | 16 | Si | Pulmón y huesos (11 meses después) |
| Medina-Franco et al. (2003) | 14 | F | NF tipo I | 2,5 | No | ExLa+RAD | No inf | No inf | No inf |
| Petros et al.8 (2005) | 25 | F | NF tipo I | 9 | No | M | No inf | No inf | No inf |
| Elsaify et al. (2006) | 18 | F | Esporádico | 4 | No | ExLa+RAD | 36 | No | No |
| Thanapaisal et al. (2006) | 19 | F | Esporádico | 7 | No | M+RAD | No inf | No inf | No inf |
| Kim et al. (2006) | 33 | F | NF tipo I | No inf | No inf | No inf | No inf | No inf | No inf |
| Dhingra et al.6 (2007) | 38 | F | Esporádico | 3 | No | ExLa | No inf | No inf | No inf |
| Woo et al.12 (2007) | 56 | F | Esporádico | 29 | No | M+LA | 6 | No | No |
| Wang et al. (2009) | 62 | Mc | Esporádico | 2,6 | No | M+RAD | No inf | No inf | No inf |
| Yi et al. (2009) | 59 | F | Esporádico | 2,5 | No | ExLa+RAD | 12 | No | No |
| Akhator et al. (2010) | 41 | F | NF tipo I | 10 | No | M+LA | 7 | Sí (9 m después) | No |
| Chalkoo et al. (2011) | 60 | F | Esporádico | 26 | No | M+LA | 12 | No | No |
| Chawla et al.9(2013) | 32 | F | Esporádico | 3,5 | No | ExLa | No inf | No inf | No inf |
| Manimozhi et al. (2015) | 40 | F | No inf | No inf | No | No inf | No inf | No inf | No inf |
| Redzepagic et al.10 (2016) | 65 | F | Esporádico | 2 | No | ExLa | 6 | No | No |
| Shuayb y Begum7 (2017) | 16 | F | Esporádico | 5 | No | M +QT +RAD | 36 | No | No |
| Hernández et al.11 (2017) | 30 | F | Esporádico | 16 | No | M | No inf | No inf | No inf |
| Caso actual (2019) | 48 | F | Esporádico | 10 | No | M+RAD | 13 | No | No |
ExLa: excisión local amplia; F: Femenino; LA: linfadenectomía axilar; M: mastectomía; Mc: masculino; NF tipo I: neurofibromatosis tipo I; No inf: no información; QT: quimioterapia; RAD: radioterapia.
En la mama el diagnóstico del TMVNP es muy difícil por su rareza y por la ausencia de características clínicas y/o radiológicas específicas12, siendo preciso un estudio histológico e inmunohistoquímico exhaustivo de la lesión que nos permita además realizar un diagnóstico diferencial adecuado con el tumor filodes maligno, el fibrosarcoma y el leiomiosarcoma6,7. El diagnóstico en material de la biopsia con aguja gruesa puede ser limitado e impreciso, aumentando de forma importante la posibilidad de diagnóstico erróneo, como ocurrió en nuestro caso, siendo necesario realizar un muestreo amplio y exhaustivo en el espécimen de resección completa.
Macroscópicamente son tumores bien circunscritos, sin llegar a estar encapsulados, y microscópicamente presentan características inespecíficas con zonas hipercelulares con células fusiforme dispuestas en fascículos que se cruzan e intercalan con áreas mixoides hipocelulares, diferenciándose de los tumores benignos de la vaina de los nervios periféricos (neurofibroma) por su elevada actividad mitótica1. A nivel inmunohistoquímico, la proteína S100 es el marcador más sensible para determinar el origen neural de las lesiones, apareciendo en el 50-90%13 de los casos dependiendo del grado de diferenciación del tumor, y el marcador mesenquimático vimentina suele ser positivo en el citoplasma de las células fusiformes. Típicamente son negativos para citoqueratinas y actina, pero la presencia de células epiteliales en algunos TMVNP puede condicionar positividad para CKAE1/AE3. Algunos TMVNP pueden mostrar positividad al CD34 indicando heterogeneidad celular, así como al CD57, LEU-7 y proteína básica de la mielina en el 50% de los casos.
El tratamiento recomendado es la exéresis quirúrgica completa de la lesión con márgenes libres y radioterapia adyuvante14 para disminuir la tasa de recurrencia local y metástasis a distancia, pudiendo estar contraindicado en el caso de tumores radioinducidos. Dado que la vía principal de diseminación es hematógena, no es preciso la disección axilar. En general su pronóstico es malo, dado que presenta un comportamiento agresivo y un alto porcentaje de recurrencias locales y a distancia; no obstante, en la literatura7-12 no hay datos disponibles sobre supervivencia del TMVNP en la mama.
En conclusión, el TMVNP primario de mama es una patología extremadamente rara con muy pocos casos reportados en la literatura, siendo necesario un análisis histopatológico e inmunohistoquímico minucioso para confirmar su diagnóstico. A pesar de la cirugía y la radioterapia adyuvante, tiene muy mal pronóstico.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

