




El síndrome de Sjögren (SS) es una enfermedad crónica autoinmune caracterizada por un infiltrado inflamatorio a nivel de las glándulas exocrinas. El SS puede ser primario o secundario (cuando aparece en asociación con otras enfermedades sistémicas). Tanto en la forma primaria como en la secundaria, la destrucción de las glándulas exocrinas conduce a un «síndrome seco», una combinación de ojo seco (xeroftalmía) y de boca seca (xerostomía). Pero el SS puede tener también manifestaciones clínicas extraglandulares (cutáneas, articulares, pulmonares, renales, gastrointestinales). Desde los años 80 se han propuesto multitud de criterios de clasificación de este síndrome. Actualmente se aceptan los criterios europeoamericanos de 2002. Según estos criterios, para el diagnóstico de SS son necesarios 4 de los 6 criterios, siendo necesario que uno de ellos sea una biopsia de glándula salival positiva o bien Ac anti-Ro/La positivos. El tratamiento abarca 2 aspectos diferentes: por un lado, el tratamiento de la sequedad de ojos y boca (agentes sustitutivos de lágrimas, fármacos agonistas muscarínicos, etc.) y por otro lado, el abordaje de las manifestaciones extraglandulares (antiinflamatorios no esteroideos, corticoides, agentes modificadores de la enfermedad, agentes citotóxicos e incluso terapias biológicas).
Sjögren's Syndrome (SS) is a chronic autoimmune disorder of the exbaocrine glands with associated lymphocytic infiltrates of the affected glands. SS occurs in a primary form not associated with other diseases and in a secondary form that complicates other rheumatic conditions. In both primary and secondary SS, decreased exocrine gland function leads to the “sicca complex”, a combination of dry eyes (xerophthalmia) and dry mouth (xerostomia). The clinical manifestations also include extraglandular disease features (skin, joints and muscles, lungs, kidneys, gastrointestinal tract). A variety of classification schemes have been proposed and discussed in the literature since the 1980s. The latest criteria, known as the American-European classification criteria for SS, were proposed in 2002. These criteria suggest that diagnosis of primary SS requires 4 of 6 criteria, including a positive minor-salivary biopsy sample and/or SS-A/SS-B antibodies. Therapy can be grouped into two separate aspects: treatment of dry eyes and dry mouth (replacement tears, muscarinic agonist agents…) and treatment of extraglandular manifestations (steroidal and non-steroidal anti-inflammatory agents, disease modifying agents, cytotoxic agents and biological therapies).
El síndrome de Sjögren (SS) es una enfermedad que se caracteriza por sequedad bucal y ocular. Las primeras descripciones de pacientes con sequedad de mucosas se realizaron a finales del siglo XIX; Mickulicz, en 1888, presentó el caso de un varón con tumefacción de las glándulas parótidas en relación con un infiltrado linfocítico. Posteriormente, Henrick Sjögren en 1933, describió a 19 mujeres con queratoconjuntivitis seca y sequedad bucal, siendo el primer autor que englobó estos hallazgos dentro de una enfermedad sistémica1. En 1960, se descubrieron los primeros autoAc implicados en el SS.
En la actualidad se sabe que el SS es una enfermedad autoinmune caracterizada por la infiltración de linfocitos T a nivel de las glándulas exocrinas. Esta infiltración origina una destrucción de las glándulas exocrinas y la aparición de sintomatología relacionada con la sequedad de las mucosas infiltradas. Además, se ha visto que hasta en un tercio de los pacientes pueden presentar diferentes manifestaciones extraglandulares, más activas y graves que las glandulares y que condicionan el pronóstico de la enfermedad a largo plazo2.
La frecuencia de este síndrome oscila entre el 0,5–3% y predomina en el sexo femenino (con una relación de 9 mujeres frente a 1 hombre)3. La edad de aparición más frecuente es en jóvenes (alrededor de los 30 años) y en mujeres posmenopáusicas. Se trata probablemente de la enfermedad autoinmune más frecuente, aunque su escasez de síntomas conlleva a que, a menudo, esté infradiagnosticada4.
El SS puede ser primario o secundario, es decir, asociado a enfermedades autoinmunes crónicas, principalmente artritis reumatoide, lupus eritematoso sistémico (LES) y esclerosis sistémica5.
EtiopatogeniaComo en la mayoría de las enfermedades autoinmunes, la etiopatogenia es multifactorial. Actualmente se acepta la teoría que explica la infiltración de glándulas salivales y lagrimales por células linfoplasmocitarias junto con la hiperestimulación de linfocitos B. La infiltración linfocitaria destruye de manera progresiva las glándulas exocrinas.
Diversos autores6–11 han planteado diferentes hipótesis que justifican la alteración de la respuesta autoinmunitaria, entre las que destacan:
- •
Alteración del reconocimiento inmunitario por la presencia de factores intrínsecos (autoantígenos) o extrínsecos (infecciones virales).
- •
Alteración de la respuesta inmunitaria adquirida por disfunción de los linfocitos B o por alteración de los linfocitos T.
- •
Alteración de la regulación de la respuesta inmunitaria por incremento del factor estimulador de células B y por alteración en la actividad de citoquinas con incremento en sangre periférica de Th2 y predominio de la respuesta Th1.
- •
Factores genéticos: entre los genes implicados en el SS destacan los haplotipos DRw52, DR2, DR3 y B8.
- •
Factores externos virales: del grupo herpes (Epstein-Barr, herpes simple 6, citomegalovirus), VHC y VHB, parvovirus B19, enterovirus (Coxsackie) y retrovirus: VIH y virus linfotrópico T humano tipo I.
- •
Autoantígenos: ribonucleoproteínas Ro/La, fodrinas y acuaporinas.
- •
Disfunción de linfocitos B: la proliferación de células B policlonal puede transformarse en bandas B oligoclonales o monoclonales.
- •
No obstante, el factor que parece desempeñar un factor clave en modificar el curso que va desde la infiltración linfocitaria inicial del tejido glandular por linfocitos T en pacientes con SS hasta el desarrollo de lesión glandular crónica es el incremento del factor activador de células B (BAFF), también conocido como estimulador de linfocitos B (Blas), que incrementa la proliferación y la supervivencia de linfocitos B y produce mayor apoptosis y destrucción glandular.
- •
Clásicamente se ha considerado que la hipofunción glandular era exclusivamente producida por la destrucción acinar secundaria a la infiltración linfocitaria; sin embargo, la hipofunción de las glándulas exocrinas5 parece ser debida también a una inhibición del proceso secretorio, producido, a su vez, por:
- •
Inhibición de la liberación de acetilcolina por citoquinas (interleuquina 1, factor de necrosis tumoral).
- •
Metabolismo aumentado de la acetilcolina (debido al aumento de la acetilcolinesterasa).
- •
Bloqueo de receptores muscarínicos M3.
- •
Alteración de la producción de óxido nítrico.
- •
Alteración de la producción de acuaporinas.
Las manifestaciones clínicas del SS se dividen en manifestaciones glandulares y extraglandulares12.
1.- Manifestaciones glandularesLa afectación de las glándulas exocrinas se manifiesta fundamentalmente por xeroftalmía, xerostomía, tumefacción de glándulas salivales mayores y afectación de otras glándulas exocrinas (a nivel cutáneo, faríngeo, etc.).
La sequedad ocular (xeroftalmía) afecta al 5–17% de población adulta5. Los signos y síntomas más frecuentes son: sequedad y disminución de lagrimeo, prurito, sensación de cuerpo extraño, hiperemia de la conjuntiva y fotofobia.
Se produce una disminución del flujo lacrimal, alteración de la composición de la lágrima, inestabilidad de la capa lacrimal (ruptura precoz) y todo ello, puede conducir a una lesión del epitelio ocular. Como consecuencia aumenta el riesgo de sufrir infecciones bacterianas y de producirse úlceras corneales.
En la sequedad oral (xerostomía) hay afectación de las glándulas salivales mayores y menores con disminución de flujo salival. Afecta a la calidad de vida de los pacientes dificultando actividades básicas como masticar, deglutir y hablar. Existe tendencia a la fisurización y ulceración de las mucosas, produciendo, como consecuencia, una intolerancia a las prótesis dentales. Además, al disminuir la capacidad antimicrobiana de la saliva, aumenta el riesgo de sufrir infecciones orales oportunistas (candidiasis oral fundamentalmente) y existe una mayor predisposición a caries13 y a enfermedad periodontal.
La tumefacción de glándulas salivales afecta al 30–50% de los pacientes. Existen 2 formas de presentación: aguda y crónica. La forma aguda se resuelve en 2–3 semanas y siempre hay que descartar sobreinfección, mientras que en la crónica se debe descartar linfoma si la tumefacción parotídea es de consistencia aumentada y se asocia a adenopatías.
La tumefacción parotídea unilateral y bilateral tiene generalmente diferentes causas. La unilateral sugiere una de las siguientes causas que pueden estar relacionadas con el SS o ser una complicación del mismo:
- •
Infección bacteriana.
- •
Litiasis.
- •
Neoplasia (adenoma, adenocarcinoma, linfoma, etc.).
Además del SS, las causas de la tumefacción parotídea bilateral pueden ser:
- •
Infección vírica (EBV, paperas, citomegalovirus, coxsackie A).
- •
Amiloidosis.
- •
Enfermedades granulomatosas (sarcoidosis, tuberculosis).
- •
VIH.
- •
Hiperlipidemia.
- •
Cirrosis/alcoholismo.
- •
Acromegalia/DM.
- •
Anorexia.
Otras manifestaciones de afectación de glándulas exocrinas son: la sequedad vaginal que conlleva dispareunia en mujeres posmenopáusicas y aumenta el riesgo de infecciones por cándida14, sequedad cutánea que predispone a prurito, sequedad nasal y faríngea que puede ocasionar reflujo gastroesofágico, tos seca crónica, rinitis no alérgica y sinusitis15.
2.- Manifestaciones extraglandularesEl 30% de los pacientes presentan este tipo de manifestaciones, entre las que se incluyen las musculoesqueléticas, neurológicas, cutáneas, renales y gastrointestinales12.
Afectación musculoesqueléticaAproximadamente el 50% de los pacientes presentan artralgias, con o sin evidencia de artritis16. Se trata de una artropatía simétrica, no erosiva que afecta principalmente a pequeñas articulaciones. El factor reumatoide puede ser positivo en el 50% de los casos17.
Son frecuentes las mialgias, aunque raramente se llega a producir una miopatía clínica y pueden existir elevaciones discretas de la creatín fosfokinasa.
Afectación neurológica y afectación psiquiátricaDentro de la afectación neurológica puede haber afectación del SNC y periférico. Cuando hay afectación del sistema nervioso periférico18 se puede producir ataxia sensitiva, polineuropatía mixta sensoriomotriz que afecta principalmente a los miembros inferiores, neuropatía autonómica, mononeuritis múltiple y afectación de pares craneales, siendo lo más frecuente la neuralgia del trigémino.
La afectación del SNC es menos frecuente (1%) y cursa con meningitis aséptica, mielitis transversa, alteraciones de la concentración y de la memoria, deterioro cognitivo y déficit motor19,20.
Por último, señalar que en estos pacientes son muy frecuentes los trastornos del ánimo21 como la depresión, ansiedad, insomnio, astenia y la fibromialgia.
Afectación cutáneaLa vasculitis cutánea ocurre aproximadamente en el 10% de los pacientes, los cuales tienen más riesgo de desarrollar manifestaciones extraglandulares, incluyendo linfoma22. La púrpura palpable es el signo más común, pero puede haber también lesiones urticariformes, máculas, pápulas y pequeñas áreas de ulceración23. Estas lesiones son más frecuentes en los miembros inferiores.
Otras manifestaciones cutáneas menos frecuentes son eritema nudoso, livedo reticularis, liquen plano, vitíligo y fenómeno de Raynaud.
Afectación gastrointestinal y hepáticaLa disfagia es un síntoma frecuente que afecta al 35% de los pacientes. También son manifestaciones frecuentes la gastritis atrófica, aclorhidria y anemia perniciosa. Se debe descartar infección por Helicobacter pylori por su asociación con linfoma MALT24. Un porcentaje importante de pacientes presenta Ac anticélulas parietales.
Es importante recordar que el SS puede ir asociado a cirrosis biliar primaria (hasta el 7% de los pacientes pueden presentar Ac antimitocondriales), enfermedad celiaca y pancreatitis autoinmune. Se debe realizar un diagnóstico diferencial con hepatitis por VHC.
Afectación renalEngloba la nefritis intersticial caracterizada por acidosis tubular renal distal tipo I (síndrome de Fanconi) que se caracteriza por la imposibilidad para acidificar la orina a pesar de existir una acidosis metabólica concomitante. Puede existir también hipopotasemia y, en casos extremos, producirse parálisis periódica hipocalémica, además del riego aumentado de cálculos renales y de osteomalacia25,26. Otras manifestaciones pueden ser la diabetes insípida nefrogénica y las glomerulonefritis (membranosa o membranoproliferativa), siendo las glomerulonefritis mucho menos frecuentes que en el LES.
Afectación pulmonarLa enfermedad pulmonar intersticial es relativamente frecuente pudiendo afectar al 25% de los pacientes27. En la mayoría de los casos, suele cursar de forma asintomática, aunque dependiendo del tipo y estadio de la enfermedad pulmonar puede producir disnea de manera progresiva, tos seca y dolor pleurítico. Incluso se han descrito casos de hipertensión pulmonar.
Existen varios tipos de enfermedad pulmonar intersticial28, entre ellos la neumonía intersticial no específica, neumonía intersticial linfoide, neumonía organizada y el pseudolinfoma (infiltrado de linfocitos maduros que no cumple criterios de malignidad).
En pacientes asintomáticos se recomienda un seguimiento semestral o anual, en el que se realizarán pruebas de de imagen y de función respiratoria.
Al inicio de la enfermedad, aunque no se observen anomalías en la Rx simple, se recomienda realizar un TAC torácico de alta resolución que podría revelar las siguientes alteraciones: imágenes de vidrio deslustrado, nódulos subpleurales, bronquiectasias y bullas. La espirometría puede poner de manifiesto un patrón restrictivo.
Afectación de tiroidesLa tiroiditis autoinmune ocurre en el 15% de los pacientes, siendo más frecuente en familiares de primer grado5.
Asociación a linfomaEl riesgo de desarrollar linfoma es del 5%, es decir, 16–44 veces mayor que en la población normal29. El tiempo medio desde el diagnóstico de SS y el del linfoma es de 6,5–7,5 años30.
Existen una serie de factores de riesgo, entre los que destacan31,32: vasculitis cutánea, neuropatía periférica, crioglobulinemia tipo II, hipertrofia parotídea/adenopatías, Ac anti-Ro/La, descenso del complemento, gammapatía monoclonal, anemia y linfopenia.
Los tipos de linfomas más frecuentes son los linfomas B de bajo grado, los linfomas MALT y los de localización extranodal (parótida, tracto gastrointestinal y pulmón). Son la causa de muerte en el 20% de los pacientes con SS.
DiagnósticoAlteraciones analíticasLa VSG está elevada en la mayoría de los pacientes (80–90%), lo que se relaciona de forma directa con una elevación importante de las proteínas circulantes, especialmente, a expensas de una marcada hipergammaglobulinemia4. Se han descrito elevaciones mayores de la VSG en aquellos pacientes Ro/La+33. Es característico, sin embargo, que la proteína C reactiva sea negativa.
En el 70% de los pacientes, las gammaglobulinas séricas están elevadas y es uno de los principales datos analíticos del SS. Asimismo, la existencia de gammapatía monoclonal es un dato frecuente (70%) y obliga a descartar su asociación con un proceso linfoproliferativo o un síndrome crioglobulinémico34.
La existencia de alteraciones hematológicas es un dato analítico que con frecuencia (40–50%) nos encontramos en estos pacientes. La anemia está presente en el 16–50%, siendo el tipo más común la normocítica normocrómica4. Otros tipos de anemia descritos son la hemolítica (suele hallarse en pacientes con SS asociado a artritis reumatoide y LES). Entre el 12–33% de los pacientes pueden presentar leucopenia35. La asociación de leucopenia con síndrome de Felty se ha descrito en enfermos con SS asociado a AR. La neutropenia y la trombocitopenia son manifestaciones poco comunes.
Finalmente, a modo de resumen, señalar que un patrón típico de presentación de SS sería VSG elevada, con proteína C reactiva negativa, hipergammaglobulinemia y FR positivo.
Alteraciones en el perfil inmunológicoLa positividad de los Ac antinucleares suele ser superior al 80%36, asociándose con mayor frecuencia con afectación pulmonar y fenómeno de Raynaud. En la mayoría de los estudios realizados, se demuestra que el FR está elevado en el 50%36 de los pacientes y se relaciona con la presencia de afectación articular, vasculitis cutánea, crioglobulinemia y Ac anti-Ro/SS-A y anti-La/SS-B.
Los Ac anti-Ro/SS-A y anti-La/SS-B se consideran los más específicos para el diagnóstico del SS aunque aparezcan en porcentajes variables (30–70%). Existe una clara relación ente los Ac anti-Ro/SS-A maternos y el bloqueo cardíaco congénito37.
En el 16% de los pacientes se detectan crioglobulinas y en el 12%, hipocomplementemia34. Además, es importante remarcar la asociación entre las crioglobulinas y la hipocomplementemia con la posible aparición de vasculitis, linfoma y VHC.
Debido a que el SS se puede asociar con otras enfermedades autoinmunes, otro tipo de Ac que se pueden detectar son los órganos específicos (anticélulas parietales, antimitocondriales, antitiroperoxidasa). Asimismo, otros autoAc que pueden aparecer son anti-ADN nativo, anti-SM, anti-RNP y anticentrómero.
Determinación de la afectación glandular1.- Estudio de la función lagrimalLa prueba más utilizada es la de Schirmer, que consiste en colocar un papel de filtro en el párpado inferior y se considera positiva cuando a los 5min la lágrima ha empapado el papel menos de 5mm.
La tinción con Rosa de Bengala, consiste en aplicar un colorante en el fórnix conjuntival inferior y visualizar, mediante una lámpara de hendidura, las posibles alteraciones en el epitelio conjuntival o corneal.
El tiempo de ruptura de la lágrima consiste en aplicar una gota de fluoresceína en el fondo conjuntival inferior y se mide el tiempo que transcurre desde el último parpadeo hasta que aparece la primera mancha oscura utilizando una lámpara de hendidura (con un filtro azul cobalto).
2.- Estudio de las glándulas salivares mayoresPara el estudio de la estructura anatómica se pueden utilizar técnicas gammagráficas, sialográficas, ecográficas y últimamente se está empezando a emplear RM.
La gammagrafía parotídea es una de las técnicas más empleadas actualmente. Permite la evaluación funcional de las glándulas salivales (valora zonas no funcionantes) y mide la asimetría entre 2 glándulas parótidas.
La sialografía nos permite ver las alteraciones en los conductos de la parótida y el aumento de sialectasias. Está técnica está en desuso por ser cruenta, por el riesgo de ruptura de ductus y por necesitar un personal altamente experimentado.
La ecografía parotídea nos proporciona una imagen anatómica y muestra áreas hipoecoicas y diversos grados de desestructuración que se podrían corresponder con focos de infiltración linfocitaria.
En la actualidad se empieza a utilizar la RM parotídea38. Es una técnica no invasiva con la que se obtienen mejores imágenes anatómicas que con la ecografía puesto que se visualizan mejor las glándulas y el conducto de Stenon y además permite diferenciar los linfomas.
La sialometría es una técnica cuantitativa que mide el flujo salivar sin estimulación (debe ser mayor a 1,5ml en 15min). Esta técnica tiene como inconveniente que el flujo salival depende de varios factores como edad, sexo, fármacos que se tomen y hora del día en que se realice la técnica.
3.- HistopatologíaLa biopsia salival permite valorar la estructura glandular y la infiltración inflamatoria. El infiltrado linfocitario está formado por linfocitos T CD4 (50%), CD8 (10–20%) y linfocitos B (20–35%), localizándose en ductos y acinos, afectando a la mayoría de las glándulas, llegando a reemplazar los acinos normales.
Uno de los criterios de clasificación del SS es la sialoadenitis linfocítica focal que requiere al menos un agregado (focus score) de un mínimo de 50linfocitos/4mm2 en áreas periductales/perivasculares.
Criterios clasificatoriosEl carácter multifactorial del SS, su tendencia a la evolución crónica y el hecho de que los síntomas iniciales sean muy inespecíficos y frecuentes en mujeres posmenopáusicas retrasan el diagnóstico de estas pacientes en varios años2.
Existe cierta controversia respecto a los criterios clasificatorios del SS. Se han propuesto multitud de criterios (Daniels, San Diego, Europeos de 1993), aunque los actualmente aceptados son los Europeosamericanos39 de 2002 (tabla 1) que presentan una sensibilidad del 90% y una especificidad del 95%; no obstante, son demasiado restrictivos ya que, según estos criterios, tiene que existir una biopsia de glándula salival patológica y/o anti-Ro/anti-La+. La biopsia de glándula salival es una prueba molesta para el paciente y puede dar falsos positivos puesto que los fumadores y las personas que toman corticoides pueden presentar alteraciones histológicas en dicha biopsia. Por otra parte, los Ac anti-Ro/anti-La pueden ser negativos en personas con pobre expresión inmunológica.
Criterios europeosamericanos (2002). Para la clasificación del síndrome de Sjögren
|
Para el diagnóstico del síndrome de Sjögren se requiere:
|
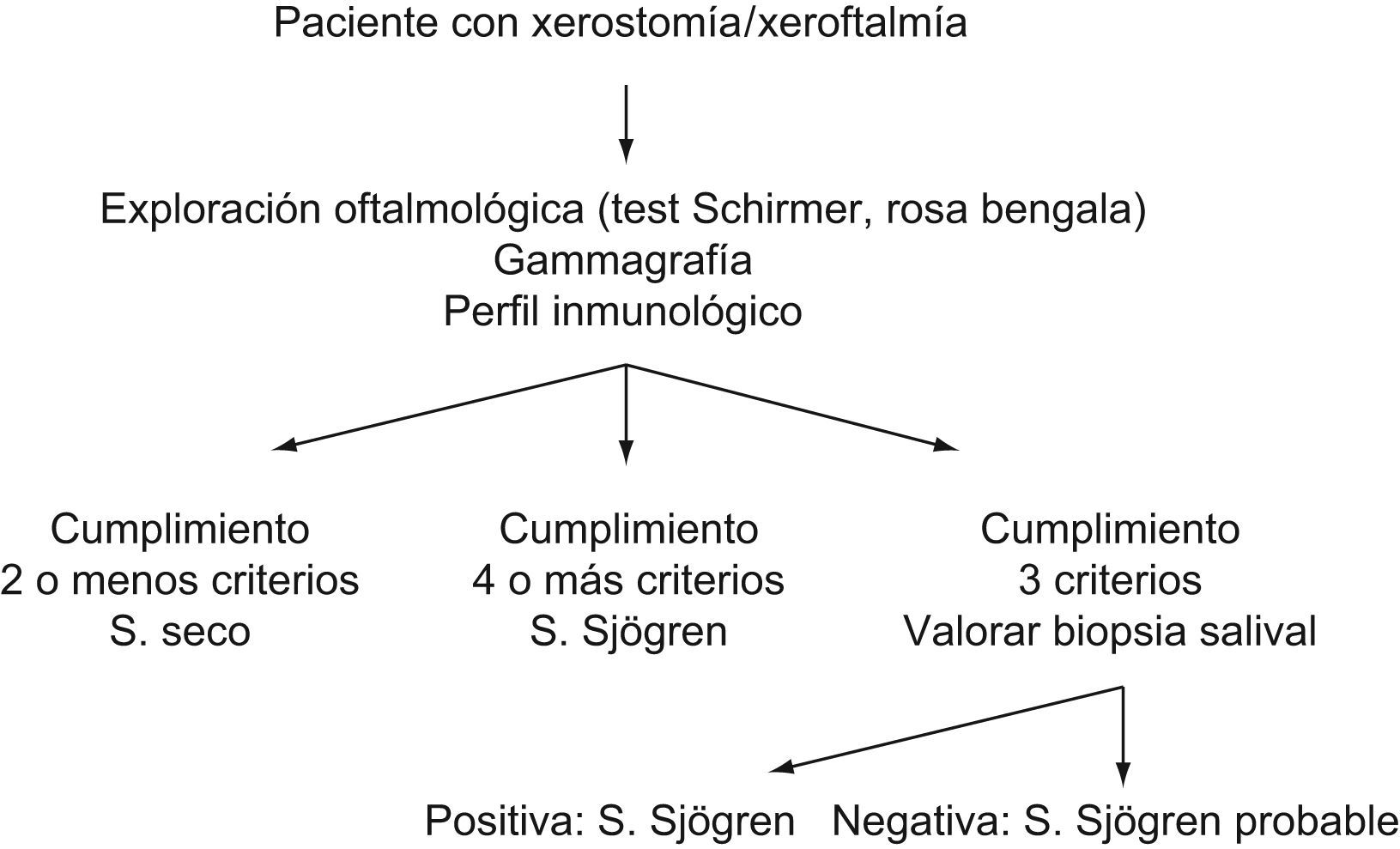
En la figura 1, se representa el algoritmo diagnóstico a seguir en un paciente con posible SS.
Diagnóstico diferencial de las manifestaciones del síndrome de Sjögren
Cuando un paciente presente sequedad ocular u oral, además de en el SS se debe pensar en otras causas (psicológicas, fármacos u otras enfermedades) que se resumen en la tabla 2.
Diagnóstico diferencial de las manifestaciones del síndrome de Sjögren
| Ejemplos | |
| Ojo seco | |
| Síndrome de Sjögren | |
| Infiltración de glándula lacrimal | Sarcoidosis |
| Amiloidosis | |
| Linfoma | |
| Psicológico | Ansiedad/depresión |
| Fármacos | Antihistamínicos |
| Antidepresivos | |
| Hepatopatías | VHC |
| Otras | Falta de vitamina A |
| Penfigoide ocular | |
| Síndrome de Stevens-Johnson | |
| Lentes de contacto | |
| Aumento de evaporación lacrimal (ordenador) | |
| Blefaritis/conjuntivitis crónica | |
| Alteración parpadeo (Parkinson) | |
| Boca seca | |
| Enfermedades sistémicas | Síndrome de Sjögren |
| Amiloidosis | |
| Sarcoidosis | |
| VIH | |
| DM | |
| Alzheimer | |
| Esclerosis múltiple | |
| Fibromialgia | |
| Fármacos | Antidepresivos |
| Antihistamínicos | |
| Anticolinérgicos | |
| Neurolépticos | |
| Diuréticos | |
| Psicológicas | Ansiedad/depresión |
| Hepatopatias | VHC |
En el diagnóstico diferencial hay que hacer mención especial a la hepatopatía crónica producida por el VHC. Los pacientes con infección por VHC pueden presentar síntomas de síndrome seco, infiltración linfocítica de glándulas salivales, auto-Ac y test oculares positivos y, sin embargo, la infección por VHC permanecer en estadio subclínico durante años y por ello ser diagnosticados erróneamente de síndrome seco40. Es importante por tanto sospechar que detrás de un síndrome seco puede haber una infección por VHC en los siguientes supuestos41:
- •
Hombres, sobre todo, de mayor edad que en el SS.
- •
Que exista una vasculitis cutánea o bien una neuropatía periférica.
- •
Se objetiven alteraciones hepáticas (clínicas o analíticas).
- •
Aparezca una neoplasia (linfoma/hepatocarcinoma).
- •
En la analítica, las crioglobulinas o el FR sean positivos y exista hipocomplementemia y los Ac anti-Ro/La sean negativos.
Debemos recordar también que el término SS secundario a VHC se debe emplear en pacientes que cumplan los criterios modificados de 2002; en caso de que no se cumplan estos criterios, se debe hablar de síndrome seco asociado a VHC.
TratamientoEl SS siempre se ha considerado una de las enfermedades autoinmunes con un menor abanico de opciones terapéuticas, sobre todo, respecto a las manifestaciones causadas por la sequedad de mucosas42. Sin embargo, en estos últimos años han aparecido nuevos fármacos que han modificado notablemente el manejo terapéutico del paciente con SS.
El SS precisa una doble aproximación terapéutica, centrada, por un lado, en el tratamiento de las manifestaciones originadas por la sequedad de las mucosas y por otro, en el tratamiento de las manifestaciones extraglandulares.
De las manifestaciones glandularesA los pacientes es necesario recomendarles medidas generales como procurar no consumir ciertos fármacos (antidepresivos, antihistamínicos, anticolinérgicos) que pueden agravar su sintomatología, evitar ambientes secos, aumentar la ingesta de agua y de productos que estimulen la secreción salival (alimentos ácidos no azucarados)43. Es importante también insistir en la necesidad de una higiene dental adecuada con visitas frecuentes al odontólogo.
Para la xerostomía se pueden aconsejar sustitutivos de saliva y sialogogos (bromexina, N-acetilcisteína), mientras que para la xeroftalmía podemos emplear lágrimas artificiales (derivados de celulosa, derivados polivinílicos, polisacáridos), colirios con mucolíticos y ciclosporina tópica al 2%.
En el caso de sequedad ocular muy intensa e incapacitante que no mejore con las medidas anteriores se podría plantear el tratamiento quirúrgico que consistiría en la oclusión de los canalículos lacrimales con materiales temporales (colágeno), o bien permanentes (silicona) o la ligadura por sutura o electrocoagulación.
Agonistas muscarínicosEn la actualidad disponemos de agonistas muscarínicos como la pilocarpina y la cevimelina que estimulan la secreción acuosa en pacientes sin gran atrofia glandular.
El clorhidrato de pilocarpina, es un agente parasimpáticomimético colinérgico, con acción agonista sobre el receptor M3 de las glándulas salivales44. Se recomiendan dosis de 5mg 4 veces al día. Los efectos secundarios principales son: cefalea, náuseas, sudoración, dolor abdominal. Está contraindicado en pacientes con asma bronquial mal controlada y glaucoma.
La cevimelina, no se comercializa en Europa y en los países en que se utiliza se recomiendan dosis 30mg cada 8h. Produce menor sudoración que la pilocarpina, pero más náuseas y diarrea45.
De las manifestaciones extraglandulares- •
Para el control de las manifestaciones articulares se emplean los AINE y la hidroxicloroquina.
- •
La vasculitis leucocitoclástica suele responder a dosis bajas de esteroides e hidroxicloroquina.
- •
La afectación intersticial pulmonar se puede tratar con esteroides asociados a inmunosupresores (azatioprina, ciclofosfamida).
- •
Para la corrección de la acidosis tubular distal se emplea el bicarbonato oral.
En la glomerulonefritis, dependiendo del resultado de la biopsia renal, se usan esteroides, ciclofosfamida en bolos o micofenolato de mofetilo.
Para corregir los síntomas del reflujo gastroesofágico, se utilizan los inhibidores de la bomba de protones y es aconsejable evitar el uso de AINES.
En los casos de afectación neurológica grave, se emplean dosis altas de esteroides y si no hay respuesta se añadirán inmunosupresores o gammaglobulina i.v. Las parestesias de la polineuropatía sensorial suelen responder a pregabalina.
Terapias biológicasLos estudios realizados con infliximab y etanercept no han obtenido eficacia significativa46–50.
Rituximab (Ac monoclonal antiCD 20): es uno de los tratamientos con mayores expectativas en pacientes con sintomatología grave51. Además ya ha demostrado eficacia en el tratamiento de linfomas B. Las indicaciones de este fármaco en el SS son:
- •
Síndrome seco severo.
- •
Disfunción de glándulas salivales.
- •
Artritis severa.
- •
Neuropatía periférica.
- •
Glomerulonefritis.
- •
Vasculitis.
- •
Crioglobulinemia.
- •
Citopenias resistentes.
- •
Linfoma de células B.
Otras terapias biológicas en estudio para el tratamiento del SS son:
Ocrelizumab (Ac humanizado anti CD 20): se cree que tendrá la misma eficacia que rituximab pero con menor inmunogenicidad.
Epratuzumab (anti CD 22).
Belimumab (antagoniza BAFF/BlyS), probablemente sea clave en el control de la hiperactividad de células B que se produce en este síndrome52.
Abatacept (proteína de fusión soluble contra ligandos CD80/86).
Por último, debido a la similitud entre la etiopatogenia del LES y el SS se debe considerar que las terapias anticitoquina como los Ac monoclonales contra IL-6, IL-10 o IFNα pueden ser de utilidad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.