







Introducción
Desde el advenimiento del síndrome de inmunodeficiencia adquirida (sida) a principios de la pasada década de los ochenta, diversas enfermedades, denominadas oportunistas, han recuperado el protagonismo perdido con la mejora sanitaria y social de la población. El nombre de leucoencefalopatía multifocal progresiva (LMP) designa una entidad definida a mediados del siglo xx e inicialmente rara y que hoy día es una complicación frecuente en portadores del virus de la inmunodeficiencia humana (VIH) y una de las enfermedades definitorias de sida.
El agente causal de la LMP es el virus JC (VJC), un poliomavirus humano que sólo produce esta enfermedad y cuya acción consiste principalmente en una desmielinización en el sistema nervioso central (SNC). La traducción clínica de las lesiones desmielinizantes es común a otras patologías, lo que obliga a un diagnóstico diferencial preciso y fiable. Si bien la certeza diagnóstica sólo viene de la mano de la anatomía patológica, la morbilidad derivada de una biopsia encefálica ha estimulado el desarrollo de técnicas moleculares, que buscan el VJC en el líquido cefalorraquídeo (LCR) y que son de parecida efectividad.
Agente causal: estructura y ciclo vital del VJC
El VJC pertenece al género Polyomavirus, escindido desde hace unos años de los Papoviridae para constituir una familia propia, la Polyomaviridae1, que acoge también a los virus BK y SV40 y a otros recientemente caracterizados2.
Este virus tiene cápside icosaédrica y un genoma circular de ADN con doble cadena. La cepa tipo se denomina Mad-1 y tiene 5.130 nucleótidos3, que se reparten en 3 regiones funcionales: codificadora temprana, codificadora tardía y reguladora. La región temprana codifica la proteína reguladora T y la t. La región tardía codifica las proteínas estructurales VP1, VP2 y VP3 y una agnoproteína o LP14. La región reguladora, situada entre las otras dos, contiene el promotorcebador, el origen de replicación y el sitio de fijación del antígeno T. El virión completo mide 45 nm.
La proteína más importante dentro de las que se transcriben antes de la replicación —proteínas tempranas— es la T. Las funciones de este antígeno son numerosas: participa en la regulación de la transcripción y en la replicación e interactúa con el ADN, el ARN y las proteínas celulares5. A la proteína t se le atribuyen funciones de inducción del crecimiento, y podría participar en la transformación tumoral6,7.
De las proteínas estructurales VP1, VP2 y VP3, la mayor es VP1, responsable de la conformación de la estructura icosaédrica y que contiene los epitopos para la inducción de anticuerpos y reconocimiento8. La agnoproteína facilitaría la llegada de VP1 al núcleo, el ensamblaje de la cápside y la propagación infecciosa de una a otra célula9, y parece que interacciona con T10.
La secuenciación genética de VJC, especialmente de VP1, ha permitido definir al menos 7 diferentes genotipos, de entre los que el más numeroso en europeos y americanos es el 1. La definición de estos genotipos supone un método para estudiar la transmisión poblacional del virus, y se ha correlacionado con su virulencia11,12.
Existen 2 posibles estados del virus: actividad/lisis celular y latencia.
Inicialmente, los viriones se unen a receptores de membrana de las células próximas y son transportados al núcleo. La transcripción del genoma viral durante la infección se divide en 2 fases: precoz (antes de la replicación del ADN) y tardía (después). El ciclo lítico comienza con la expresión de la proteína temprana T13. T se fija al origen de replicación, anula la represión de ciertas enzimas de la célula e interactúa con la ADN polimerasa del hospedador, y por tanto da comienzo a la replicación viral. También activa la transcripción de los genes tardíos, que formarán la cápside. La acumulación de muchos viriones completos en el núcleo de la célula infectada causa su muerte por lisis de membrana.
La replicación de VJC se controla primariamente en la transcripción. La transcripción de las proteínas tempranas, entre ellas T, está dirigida por un conjunto de factores celulares que interaccionan con su promotor en la región reguladora, entre ellos Tst-1, NF-1, Sp1, GBPi, NF-κB, YB-1, Pur-α y GF-1, responsables del tropismo hacia las células gliales cerebrales humanas, únicas en las que puede replicarse productivamente el virus4,5,14-30. Los sitios más importantes de fijación de estos factores son 2 repeticiones en tándem de 98 pares de bases y un pentanucleótido repetido AGGGAAGGGA. Al comparar la expresión de VJC en células gliales y no gliales, la actividad del promotor vinculado a la síntesis de T era mucho mayor en las primeras15,31,32, y las otras no podían por ello albergar replicación viral, producción de cápside o padecer infección lítica.
Según la estructura de la región reguladora, las líneas de VJC se dividen en arquetípicas y reordenadas33. Se cree que los arquetipos son los responsables de la propagación entre la población y de la infección latente, y son incapaces de replicarse de manera efectiva en células gliales34-46.
El ciclo lítico del virus conduce a la destrucción del oligodendrocito, célula productora de mielinización de axones en el SNC. Cuando se afecta un número crítico de ellos se produce una lesión desmielinizante: ésta es la base patogénica de la LMP.
Sin embargo, otras posibilidades completan el abanico de acciones nocivas de este polioma. El concepto de latencia, en el cual no hay lisis del hospedador, está relacionado con el desarrollo de tumores. Hasta la fecha no se ha demostrado concluyentemente la implicación de este polioma en la carcinogénesis humana47, pero sí se han descrito casos de neoplasias gliales con detección de VJC o su antígeno T en seres humanos48-51, aunque sin confirmarse la relación de causalidad. En animales de experimentación pueden inducirse tumores neuroectodérmicos cuando se les inocula el VJC6,52.
En estos casos se produce T sin haber ciclo lítico, para que la célula infectada no sea destruida, sino inmortalizada53-55. El antígeno T interacciona con proteínas supresoras de tumores del hospedador para habilitar un microambiente óptimo que propicie la replicación viral. Ejemplo de esto es la relación con la proteína p53, que altera la intervención de ésta sobre el ADN celular anómalo56-59. Asimismo, T también genera aberraciones cromosómicas en células de cultivo60-63.
La latencia de la infección se ha confirmado mediante detección del VJC en riñón y orina, y de anticuerpos en orina, entre otros35,36,64-72. Los sitios de probable latencia son las células renales y los linfocitos B73,74, y hay indicios de VJC en orina de trasplantados renales, embarazadas y sujetos sin inmunodepresión, aparte de en enfermos de LMP.
Respecto a la latencia en el SNC, las investigaciones son contradictorias: en unas se detecta el virus sólo en tejido cerebral de pacientes con LMP75-81, y en otras, también en sanos82-87. En suma, es poco habitual el hallazgo del virus en SNC de individuos sin LMP88.
La enfermedad: leucoencefalopatía multifocal progresiva
En 1952 se observaron por vez primera los síntomas de una nueva enfermedad descrita años después por Astrom y Richardson89,90, la LMP. Los primeros casos correspondieron a 2 ancianas con leucemia linfática crónica y un síndrome neurológico rápidamente progresivo91. En 1961 Richardson sugirió una infección viral del SNC en inmunodeficientes como la causa de esta enfermedad92. Zu Rhein y Silverman encontraron en el núcleo de oligodendrocitos de los pacientes inclusiones cristalinas idénticas al virus polioma murino93,94. Padgett et al95 cultivaron en 1971 un virus similar a los papova a partir del cerebro de un enfermo de linfoma de Hodgkin con LMP, y lo llamaron "JC" por ser las iniciales del paciente.
Epidemiología
La distribución de la enfermedad ha variado sustancialmente desde entonces, sobre todo bajo la influencia de la epidemia del sida. El primer caso de LMP en un enfermo de sida se comunicó en 1982. Si antes los trastornos linfoproliferativos constituían la principal patología subyacente a la LMP, en 1991 el sida era la enfermedad de base en el 72% de los casos96. La prevalencia de LMP en el sida se sitúa entre el 1 y el 10%, y hay una incidencia de 5,7 casos por 10 millones de habitantes y año96,97, con un acusado predominio de varones y una media de edad de 38 años98.
Al margen del sida, otras enfermedades que alteran la inmunidad mediada por células, las autoinmunes (incluidas las conectivopatías) y la inmunodepresión iatrogénica, especialmente en los trasplantados99,100, constituyen patologías de riesgo para la LMP (tabla 1). En los últimos años, una terapia emergente, los anticuerpos monoclonales —utilizados en la lucha contra enfermedades de base inmune, como esclerosis múltiple, enfermedad de Crohn o artritis reumatoide— se suponen responsables de la aparición de la LMP como efecto secundario en un porcentaje escaso de tratados101-103. El más conocido es el natalizumab101, pero también se ha descrito LMP con etanercept104 y rituximab105,106.

Historia natural y manifestaciones anatomopatológicas
La distribución de la infección por VJC es universal, y el reservorio, exclusivamente humano107. La mayoría de la población se infecta en algún momento de su vida. A los 5 años de edad, al menos el 10% tiene anticuerpos anti VJC, y a los 14, la serología es positiva en el 65%. Se acepta que, como mínimo, el 75% de adultos tiene anticuerpos contra VJC, sin que manifiesten enfermedad. La mayor parte de los enfermos de LMP tenía anticuerpos previamente y éstos no se incrementan al aparecer los síntomas.
Se admite que el virus se establece en el ser humano mediante una infección latente, y que la LMP surgiría como reactivación de la infección74,108. Se desconocen la puerta de entrada del virus y los sitios de replicación primaria93,109,110, pero se especula con que el tejido amigdalar, estrechamente relacionado con una posible transmisión respiratoria y en el cual se ha encontrado VJC, podría ser el origen de la infección111,112, desde donde el virus se transferiría a linfocitos B y llegaría al riñón113,114. Los linfocitos B activados, en el contexto de unahdeficiencia inmune, pueden cruzar la barrera hematoencefálica y transmitir el virus a los oligodendrocitos encefálicos73,115,116. En más del 95% de pacientes con LMP hay linfocitos periféricos infectados.
Aunque el riñón y los linfocitos B puedan albergar una infección latente, la replicación efectiva de VJC solamente ocurre en células gliales. La alteración de la inmunidad celular es el factor esencial en la patogenia de la infección activa, mientras que la inmunidad humoral está aceptablemente representada por la persistencia de anticuerpos en los pacientes.
La principal célula afectada por la reactivación viral es el oligodendrocito. Su destrucción masiva genera lesiones desmielinizantes, donde no hay vainas de mielina pero los axones están relativamente preservados89,117. La muerte del oligodendrocito se cree que se produce por apoptosis. Los oligodendrocitos enfermos y todavía no destruidos están agrandados y presentan núcleos hipercromáticos con inclusiones de VJC118, y tienden a ocupar la periferia de las lesiones.
Los astrocitos, otras células gliales, se afectan secundariamente y exhiben morfología anómala, gran tamaño y multinucleación, con núcleos heterocromáticos y mitosis anormales89. En ellos ocurriría una infección abortiva relacionada con la aparición de neoplasias, bien porque los oligodendrocitos se resistan al proceso de transformación o por una mayor interacción con los factores supresores de tumores por parte del astrocito56,57,119. De hecho, los tumores inducidos experimentalmente por poliomas son de estirpe astrocitaria, y los astrocitos no experimentan degeneración apoptótica120.
Manifestaciones clínicas
La afectación del SNC en la LMP es de naturaleza multifocal y guarda relación con la ubicación de las lesiones desmielinizantes. Los 3 pilares clásicos de la semiología son déficit motor, deterioro cognitivo y alteración visual, y se ha señalado al primero como el más prevalente121-123.
La enfermedad puede presentarse de manera insidiosa o abrupta. El comienzo subagudo es frecuente en forma de deterioro cognitivo y demenciación. El comienzo brusco o ictal suele manifestarse como un defecto campimétrico visual o una paresia motora de miembros.
La demencia traduce por lo común una afectación cerebral extensa y subcortical, expresada como lentificación y trastorno del comportamiento, en oposición a la demencia cortical de la enfermedad de Alzheimer.
Los déficit focales, como hemiparesia y hemihipoestesia, se deben a lesiones en la corteza cerebral o subcorticales. El defecto visual traduce la afectación del córtex occipital o de las vías temporooccipitales (tabla 2).

Las lesiones del tronco encefálico y del cerebelo ocasionan defectos focales bilaterales, paresias cruzadas, afectación de pares craneales y alteración del equilibrio, de las funciones vegetativas o de la conciencia.
Las crisis epilépticas, mucho menos frecuentes y que tienen por definición un origen cortical, pueden aparecer en una enfermedad de sustancia blanca como la LMP porque al propagarse interesan a conexiones subcorticales.
Pronóstico
El curso de la LMP es habitualmente subagudo y conduce al fallecimiento entre 1 mes y 1 año del comienzo de los síntomas90,117,124. No obstante, hay enfermos con supervivencias inexplicablemente prolongadas117,125-127. Estudios de supervivientes a largo plazo indican que la actividad de linfocitos T citotóxicos dirigidos contra VJC se relaciona con una detención en la progresión de la LMP128,129. Hay asimismo constancia de remisiones completas, espontáneas o por la retirada de medicación inmunosupresora98. En la LMP asociada a sida son de mejor pronóstico el recuento elevado de linfocitos T CD4, el tratamiento con terapia antirretroviral de alta eficacia (HAART), la carga viral reducida de VIH, el inicio del sida como LMP, la carga viral escasa de VJC en el LCR, la ausencia de efecto de masa radiológico y de progresión neurológica a los 2 meses de tratamiento y, con discrepancias, las lesiones captadoras de contraste en la tomografía computarizada (TC) o la resonancia magnética (RM)129-137.
Mientras que la introducción de la HAART en el tratamiento del sida ha conseguido disminuir la tasa de enfermedades oportunistas neurológicas como el linfoma del SNC, la encefalitis toxoplásmica, la encefalopatía por VIH, la meningitis criptocócica o la polineuropatía asociada al VIH138, no ha variado las cifras de LMP, que incluso se han incrementado138-142, aunque sí ha mejorado el pronóstico de la LMP ya declarada143.
Diagnóstico
En la práctica clínica, un paciente inmunodeficiente con sintomatología neurológica multifocal subaguda y no regresiva, tras descartar otros agentes, en ausencia de fiebre y de infección piógena y con una neuroimagen típica, tiene una alta probabilidad de padecer LMP. Las técnicas utilizadas en el diagnóstico de esta enfermedad son:
Radiología
El diagnóstico puede efectuarse por TC o por RM cerebrales, si bien ambas son inespecíficas. La RM es más sensible y puede detectar lesiones ausentes de la TC, especialmente las pequeñas, las recientes o las de la fosa posterior.
Las lesiones típicas de la LMP son bilaterales en el 75% de los casos, y se localizan frecuentemente en la sustancia blanca de alrededor de los ventrículos cerebrales o en la subcortical, especialmente en la unión córtico-subcortical y en los lóbulos parietal, occipital y frontal. Se afectan menos el córtex, los ganglios basales, el cerebelo y el tronco cerebral, y menos aún la médula espinal144,145. Los casos de LMP asociada al sida se caracterizan por la participación de las estructuras de fosa posterior, los ganglios basales y los lóbulos temporales, topografías inusuales en la enfermedad146,147.
Las lesiones son inicialmente asimétricas, y al evolucionar la enfermedad tienden a confluir hasta dibujar una disposición simétrica7. En la TC y en la secuencia T1 de la RM están hipoatenuadas, y en secuencias FLAIR y T2 de la RM son hiperintensas (figs. 1 y 2). No suele haber desplazamiento de estructuras adyacentes ("efecto de masa") ni realce con el contraste intravenoso148 (fig. 3).
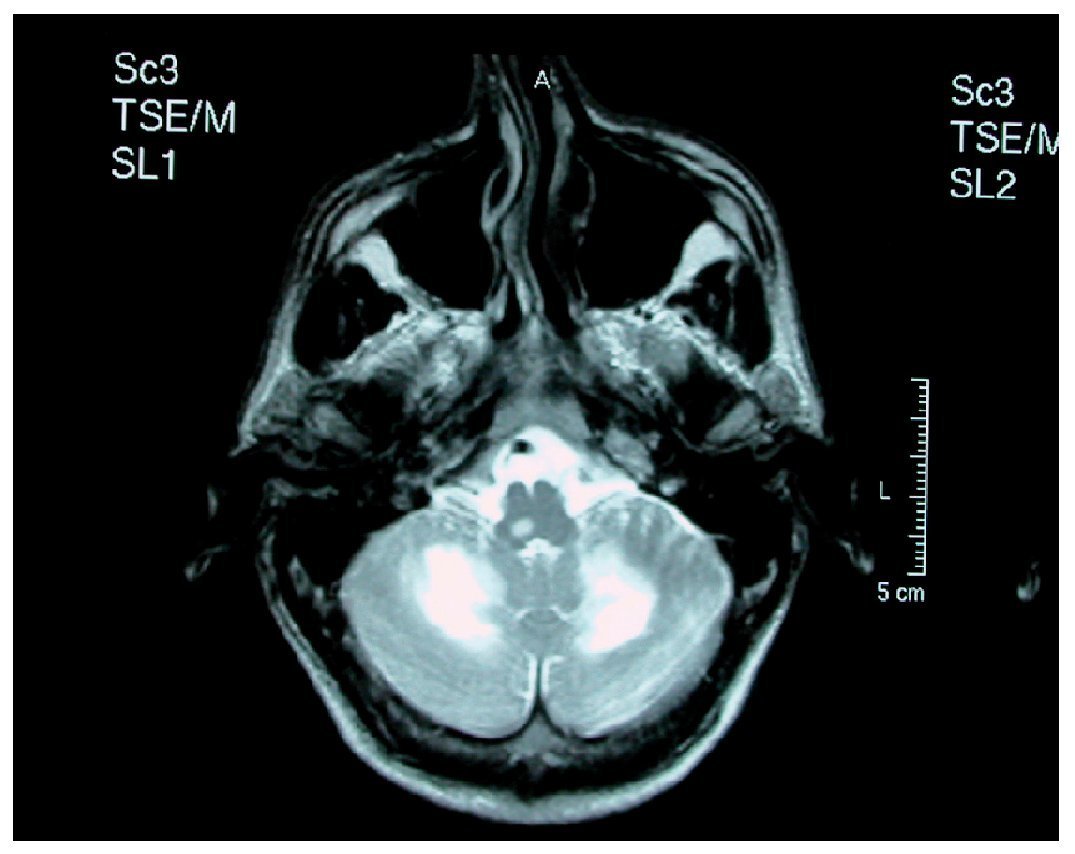
Figura 1. Resonancia magnética craneal, secuencia T2 de un paciente con leucoencefalopatía multifocal progresiva. Afectación cerebelosa bilateral y de bulbo raquídeo. Original.
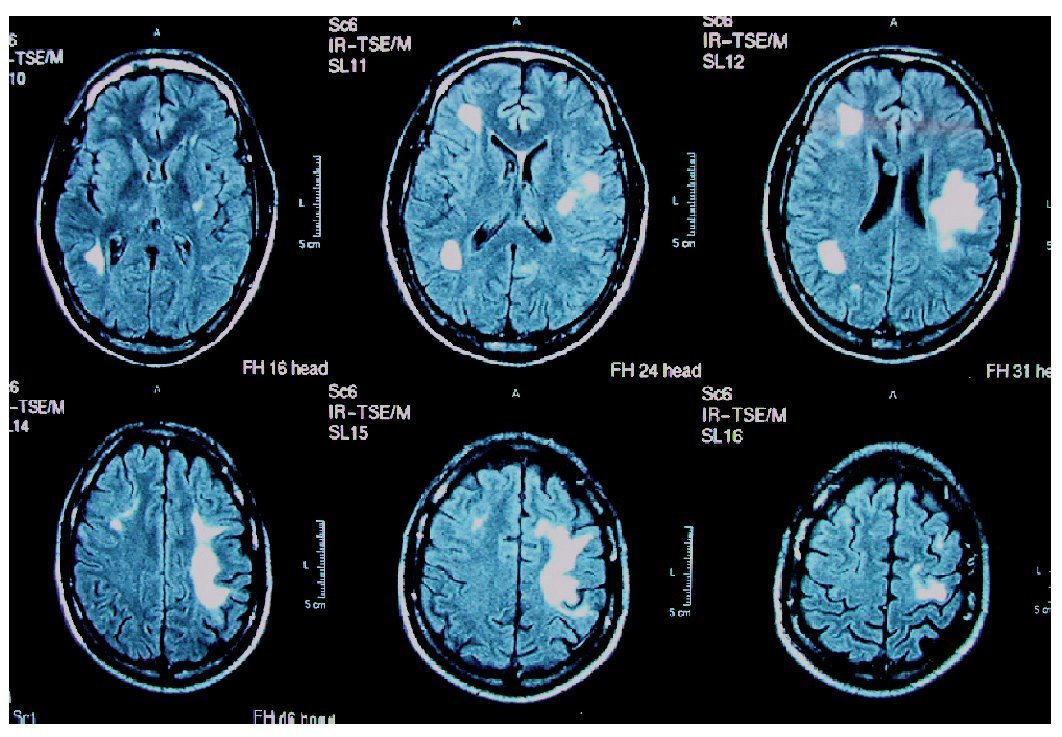
Figura 2. Resonancia magnética craneal, secuencia FLAIR de un paciente con leucoencefalopatía multifocal progresiva. Afectación frontal y parietal bilateral y asimétrica. Original.
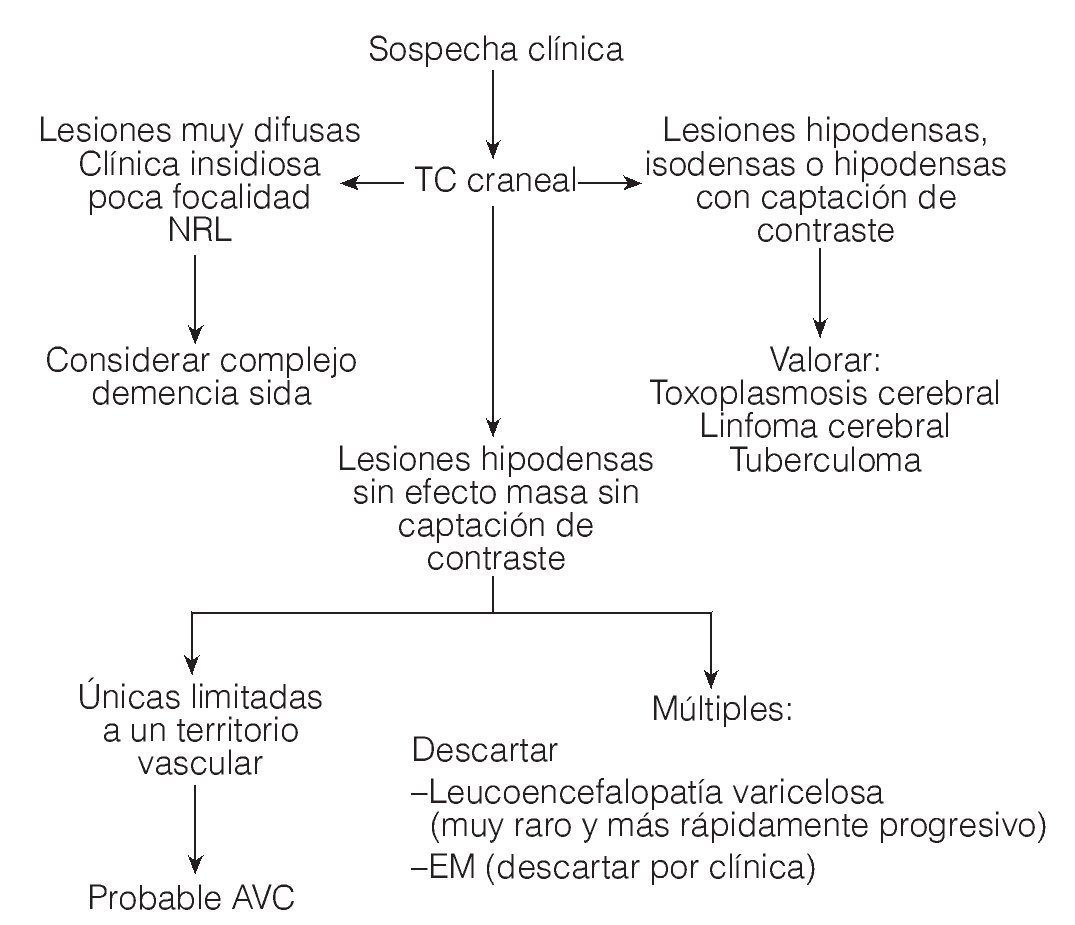
Figura 3. Algoritmo para el diagnóstico diferencial de la leucoencefalopatía multifocal progresiva mediante TC craneal. TC: tomografía computarizada; AVC: accidente vascular cerebral. (Tomada de Abós .)
Histología
Si la clínica o la radiología son insuficientes para el diagnóstico de LMP, se puede recurrir a la histología. La biopsia cerebral es, hasta la fecha, el único método de certeza para LMP135, y se prefieren las técnicas de estereotaxia por su menor morbilidad149.
Análisis molecular
Constituye hoy el método de mayor fiabilidad —al margen de la anatomía patológica— y el de mayor potencial futuro. La técnica más estudiada y de mejores resultados es la reacción en cadena de la polimerasa (PCR), que detecta el ADN del VJC en el LCR en los casos de LMP mediante amplificaciones sucesivas121,137,150-157. Actualmente se acepta un diagnóstico de LMP basado en la sintomatología, la neuroimagen y la PCR, sin necesidad de biopsia, aunque no constituye una técnica de certeza como ésta158-160.
Otras técnicas
Determinaciones de anticuerpos frente a VJC, análisis bioquímico de LCR y detección del virus en orina son poco útiles, y la viruria se ha demostrado en inmunodeficientes sin LMP, en gestantes e incluso en sujetos sanos43,161,162. La tomografía por emisión de positrones (PET) podría diferenciar la LMP del linfoma, si bien se han referido resultados contradictorios y es una técnica poco accesible163.
Diagnóstico diferencial
Diversas enfermedades plantean dificultades de diferenciación con la LMP:
Toxoplasmosis cerebral
Es la enfermedad más prevalente en el enfermo de sida con síntomas neurológicos, pero últimamente está en regresión164,165. También es la causa más frecuente de lesiones expansivas intracraneales en estos pacientes. Produce cefalea, estado confusional, déficit focales, fiebre y crisis epilépticas166,167. A diferencia de la LMP, las lesiones se realzan anularmente con el contraste y tienen efecto de masa, y hay muy buena respuesta a los antitoxoplásmicos.
Linfoma cerebral primario
Es la segunda causa más frecuente de enfermedad focal cerebral en enfermos de sida135. Se distingue de la LMP por la captación de contraste y el efecto de masa de las lesiones radiológicas, los síntomas generales como fiebre, sudoración y pérdida de peso, y la mayor prevalencia de comicialidad.
Encefalopatía por virus de la inmunodeficiencia humana
Se caracteriza por demencia subaguda, trastorno de la marcha y alteración de la conducta. Hay menor tendencia a la focalidad en la neuroimagen y en la exploración clínica que en la LMP.
Enfermedad por citomegalovirus
Afecta también al sistema nervioso periférico. Se manifiesta como encefalitis o ventriculoencefalitis. La participación extracraneal y el trastorno de la conciencia delimitan las diferencias con la LMP.
Tuberculosis del sistema nervioso central
Se presenta como meningitis, tuberculoma o absceso cerebral; puede haber hipercaptación de meninges o realce de las lesiones con el contraste radiológico. El diagnóstico se basa en el análisis del LCR, y es definitiva la presencia del bacilo de Koch en su cultivo.
Enfermedad del sistema nervioso central sin infección por el virus de la inmunodeficiencia humana
La más prevalente es la enfermedad desmielinizante primaria, sobre todo la esclerosis múltiple. La evolución de esta patología es crónica, y suele consistir en brotes focales total o parcialmente reversibles con esteroides, y la autonomía del enfermo no suele verse comprometida hasta fases avanzadas. Las lesiones radiológicas son más pequeñas y de contornos más definidos que la LMP, y tienden a situarse periventricularmente. El hallazgo de bandas oligoclonales de inmunoglobulinas en el LCR es de gran importancia en el diagnóstico.
Tratamiento de la leucoenfalopatía multifocal progresiva
La importancia del correcto diagnóstico de LMP se debe a que permite el establecimiento de un pronóstico vital y la exclusión de otras entidades semejantes semiológicamente pero con un tratamiento eficaz, del que carece la LMP.
Se han ensayado numerosos fármacos, sin claro beneficio con ninguno. Los más conocidos fueron el arabinósido de citosina y el cidofovir, de resultados controvertidos122,168-173.
Muchos estudios han relatado posteriormente supervivencias aumentadas en pacientes con VIH y LMP tratados con HAART143,174-183. Se supone que la terapia HAART produce una restauración inmune, pues incrementa el recuento de linfocitos CD4 y disminuye la presencia de VJC en el LCR178,183,184. Por ello, ésta se considera la mejor alternativa farmacológica en la LMP. Sin embargo, se ha observado también ausencia de mejoría neurológica a pesar de la buena respuesta inmunovirológica al tratamiento, e incluso se ha comunicado progresión clínica y radiológica, supuestamente por el aumento de respuesta inflamatoria en los buenos respondedores: es el "síndrome de reconstitución inmune"183,185-189.
Se ha postulado que el cidofovir sería la opción más razonable si no hay mejoría o tolerancia de la terapia HAART190.
Infección conjunta por virus de la inmunodeficiencia humana y virus JC
La comorbilidad de las infecciones por VIH y VJC determina cambios sutiles en el patrón de la LMP, que comprenden variaciones demográficas (disminución de la edad media de los pacientes y aumento de la proporción de varones), macroscópicas (con mayor implicación de la fosa posterior) y microscópicas (con frecuentes infiltrados inflamatorios perivasculares)146,147,191-194.
La mayoría de las investigaciones sobre el vínculo molecular de ambos virus tienen como diana a la proteína tat del VIH-1, que actuaría favoreciendo la síntesis de proteínas estructurales de VJC al interactuar con su promotor tardío195-200.
La infección por el VIH-1 puede, por otra parte, provocar la disrupción de la barrera hematoencefálica y facilitar el acceso al cerebro de linfocitos B infectados por el VJC201-203.
* Autor para correspondencia.
Correo electrónico:mbgg@comcadiz.com (M.B. Gómez González).
INFORMACIÓN DEL ARTÍCULO
Historia del artículo:
Recibido el 4 de febrero de 2009
Aceptado el 2 de marzo de 2009