






La miositis con cuerpos de inclusión (MCI) esporádica forma parte de las miopatías inflamatorias idiopáticas pero tiene unas características clínicas e histológicas bien definidas, aunque no siempre fácilmente identificables. Afecta mayoritariamente a varones de más de 50 años de edad, con un patrón clínico que incluye debilidad muscular y atrofia, a menudo de distribución asimétrica, lo cual implica un amplio diagnóstico diferencial. En un cierto porcentaje de casos se asocia a enfermedades autoinmunes sistémicas. No se admite, por el contrario, su asociación a neoplasia, a diferencia de lo que ocurre con las dermatomiositis. Para su diagnóstico se han propuesto criterios que combinan datos clínicos con datos histopatológicos, y las pruebas de laboratorio y la EMG son poco útiles para el diagnóstico. La resonancia magnética, en cambio, parece que puede ser de ayuda y quizá deba incluirse entre los criterios diagnósticos. La patogenia de la MCI, compleja y multifactorial, no es bien conocida, y en ella se interrelacionan fenómenos inflamatorios, mitocondriales y degenerativos. El retraso en el diagnóstico es la regla, y por desgracia la respuesta al tratamiento es, en la mayoría de los pacientes, descorazonadora, a excepción de los casos en que la MCI se asocia a una enfermedad autoinmune.
Sporadic inclusion body myositis (IBM) is a major subgroup among idiopathic inflammatory myopathies. The clinical and pathologic findings of this disease are well defined but are not always easy to identify. IBM mainly affects men aged more than 50 years old who usually present with chronic and sometimes asymmetrical weakness and atrophy, thus requiring a wide differential diagnosis. Some well-characterized autoimmune diseases are associated with IBM. However, unlike dermatomyositis, there is no association with neoplastic disease. Clinical and histopathological data are mandatory in the diagnosis of IBM, while laboratory and electromyographic studies are usually non-diagnostic. In contrast, magnetic resonance imaging may help in diagnosis and should possibly be included in the diagnostic criteria. The pathogenesis of IBM is still not well-defined, as it involves interrelations among inflammatory, degenerative and mitochondrial phenomena. Diagnostic delay is the rule, and the response to available treatments is poor except when an autoimmune disease is associated with IBM.
La miositis con cuerpos de inclusión (MCI) en su forma esporádica fue descrita como entidad patológica en 1971 por Yunis y Samaha1 y forma parte del grupo de miopatías inflamatorias idiopáticas (MII) junto a la dermatomiositis (DM), la polimiositis (PM) y la miopatía necrosante inmunomediada o autoinmune (MNA)2. Las MII constituyen un grupo heterogéneo de miopatías adquiridas que comparten la presencia de debilidad muscular, de aparición aguda, subaguda o crónica, y signos histológicos de inflamación3. Esto último, la presencia de infiltrado inflamatorio en la biopsia muscular, es la alteración histológica más relevante que diferencia la MCI esporádica de su forma hereditaria4.
A diferencia del resto de las MII, la MCI tiene predilección por el sexo masculino y la edad avanzada, y es la miopatía adquirida más frecuente en mayores de 50 años5. Su etiología es desconocida y su patogenia muy compleja, combinando fenómenos inflamatorios inmunomediados, degenerativos y mitocondriales6–11.
Clínicamente se caracteriza por debilidad muscular con frecuencia asimétrica, preferentemente distal en las extremidades superiores y proximal en las inferiores, pudiendo asociar además trastorno de la deglución. Progresa lentamente produciendo marcada morbilidad y dependencia funcional, confinando a los pacientes a utilizar silla de ruedas aproximadamente a los 10 años del inicio de los síntomas8.
Desafortunadamente, la gran mayoría de las veces la MCI es refractaria al tratamiento, por lo que el desarrollo de nuevas terapias basadas en la profundización de sus mecanismos patogenéticos es primordial para mejorar la calidad y la esperanza de vida de estos pacientes6,12,13.
EtiologíaEn 1967, Chou14 describió el que probablemente sería el primer caso documentado de MCI y, años después, Yunis y Samaha1 introdujeron el término de MCI al realizar su descripción como entidad nosológica. Se estima que la MCI representa el 16-28% de los pacientes con MII y que tiene una prevalencia global de entre 4,5-9,5 por millón de habitantes, llegando a 35 por millón en mayores de 50 años5,15,16, si bien estas cifras pueden estar infraestimadas por diagnósticos erróneos de PM en pacientes con MCI17. A pesar de haber pocos estudios poblacionales, los existentes muestran una prevalencia diferente en función de los países y la etnia, situación que queda reflejada en la tabla 1. Teniendo en cuenta estos diferentes datos poblacionales, serían necesarios estudios multicéntricos con mayor número de pacientes para definir mejor la epidemiología de la MCI y determinar su relación con factores genéticos y medioambientales.
La evidencia de una susceptibilidad genética en la MCI asienta en estudios del HLA y del complejo de histocompatibilidad mayor (CHM). La asociación con HLA-DR3 es la más sólida de las asociaciones de entidades con HLA, presente aproximadamente en el 75% de los casos, aunque varía en función de las diferentes etnias18. También se ha demostrado que el haplotipo ancestral 8.1 del CHM se asocia con MCI en australianos, holandeses y caucásicos de Estados Unidos6, así como el haplotipo 52.1 en japoneses3.
Hoy en día la etiopatogenia de la MCI continúa sin aclararse, a pesar de los numerosos estudios e hipótesis al respecto. Lo que sí parece claro es que son varios los mecanismos implicados, agrupados en inflamatorios de tipo autoinmunitario, degenerativos y mitocondriales, modulados genéticamente y posiblemente desencadenados por un estímulo que podría ser infeccioso, si bien el orden de actuación o de predominancia de cada uno de ellos está por aclarar19.
Fenómeno inflamatorioAl igual que en la PM, en la MCI se ha constatado una citotoxicidad directa mediada por citocinas (IL-1, IL-6, IL-10, TNF-α, INF-γ, STAT, TGF-β) y quimiocinas (MCP-1, PIP-1α, IP-10) inflamatorias, y predominantemente por linfocitos T CD8+ que invaden las fibras musculares que expresan antígenos del CHM-I20,21, ICAM-I, CD45RO y co-estimulador inducible (ICOS)22 en su superficie, induciendo su necrosis principalmente tras la liberación de perforinas, granzima A y granulisina23,24. Dicha invasión tiene lugar en fases iniciales de la enfermedad y su presencia es mayor que las fibras musculares con material rojo Congo positivas25, lo que en consecuencia sugiere que los fenómenos inflamatorios preceden a los degenerativos. Además, existe una respuesta aumentada de chaperonas (sobre todo Grp78) del retículo endoplásmico, resultado de la alteración en la homeostasis celular26–29, que estimula la producción de factor nuclear κβ, que a su vez también induce la liberación de citocinas inflamatorias, la expresión de antígenos de clase I del CHM y la producción de proteína precursora amiloide y otras proteínas de depósito30, dando lugar, esto último, a los fenómenos degenerativos de forma secundaria.
Por otra parte, durante mucho tiempo se había asumido la no participación de las células B en la patogenia de la MCI31,32, hasta que en estudios con microarrays sobre biopsias musculares publicados en 200233 se pudo demostrar una notoria transcripción de inmunoglobulinas, permitiendo la confirmación posterior de su participación al demostrarse la secreción de inmunoglobulinas y su expansión clonal34,35.
Todos estos mecanismos inflamatorios obligan además a sospechar la existencia de una noxa, posiblemente infecciosa, como desencadenante inicial de la enfermedad28,36, lo cual puede venir apoyado por la presencia de cambios histológicos similares entre la MCI y algunas miositis víricas37,38.
Fenómeno degenerativoEn contra de lo expuesto previamente, algunos autores defienden la patogenia de la MCI como eminentemente degenerativa. Esto se basa en la detección por inmunohistoquímica en biopsias musculares de múltiples proteínas asociadas a otros conocidos procesos degenerativos, como la proteína beta amiloide y su precursor, la proteína priónica, la apolipoproteína, la antiquimiotripsina 1, la ubiquitina, etc.39–46. Tras una producción y depósito anormal de dichas proteínas, sobre todo la precursora de beta amiloide, y su posterior fallo en la eliminación, tiene lugar un efecto tóxico directo, lo que lleva a una situación de estrés celular y del retículo endoplasmático, y de esta manera provoca una respuesta inflamatoria secundaria43. El principal punto débil de esta teoría eminentemente degenerativa es la detección de dichas proteínas de depósito también en otras miopatías inflamatorias como la DM y la PM, en ocasiones en mayor cantidad que en la MCI33,45.
Fenómeno mitocondrialSe han encontrado incrementos de los marcadores de estrés oxidativo en músculos de pacientes afectados de MCI y disminución de actividad de catepsinas D y B, supuestamente por estrés del retículo sarcoplásmico47. Se cree que distintos factores, como las variaciones genéticas en los componentes de la cadena respiratoria mitocondrial, pueden contribuir al estrés oxidativo48. El patrón de deficiencia parcial de COX en las biopsias musculares de los pacientes con MCI es similar al que se observa en la edad avanzada, aunque a menudo es de mayor intensidad. Es más, los cambios morfológicos mitocondriales en las biopsias musculares de la MCI son en ocasiones más notorios que los existentes en las verdaderas miopatías mitocondriales primarias48,49. El significado clínico de las alteraciones mitocondriales en la MCI no es bien conocido, ya que en los casos en que el metabolismo oxidativo muscular se ha estudiado mediante espectroscopia con 31P no se ha detectado ninguna alteración50. Por el contrario, en 2006, Olfords et al.51 propusieron que la disfunción mitocondrial de la MCI puede contribuir a la debilidad muscular y a la atrofia que presentan los pacientes, sugiriendo en consecuencia que sería lícito intentar aproximaciones terapéuticas destinadas a mejorar la función mitocondrial. Más recientemente, Varadhachary et al.11 analizaron con cierta profundidad el binomio «patología mitocondrial» y «miopatías inflamatorias», resaltando la carencia de estudios con series amplias (a excepción de nuestro trabajo52) e introduciendo un concepto novedoso como es el de las PM con cambios mitocondriales (PM-mito). En efecto, se trata de casos de PM que no reúnen criterios de MCI pero en los que se aprecian cambios histológicos mitocondriales, señalando un peor pronóstico respecto a los casos de PM sin cambios mitocondriales. La detección de trastornos mitocondriales requiere, como prueba diagnóstica, la realización de una biopsia muscular, si bien muy recientemente se ha descrito un eventual biomarcador sérico como es el factor de crecimiento 21 del fibroblasto (FGF-21) que se asocia a patología muscular mitocondrial. Aunque no es la casuística predominante en este trabajo, sí se incluyen algunos casos de MCI53.
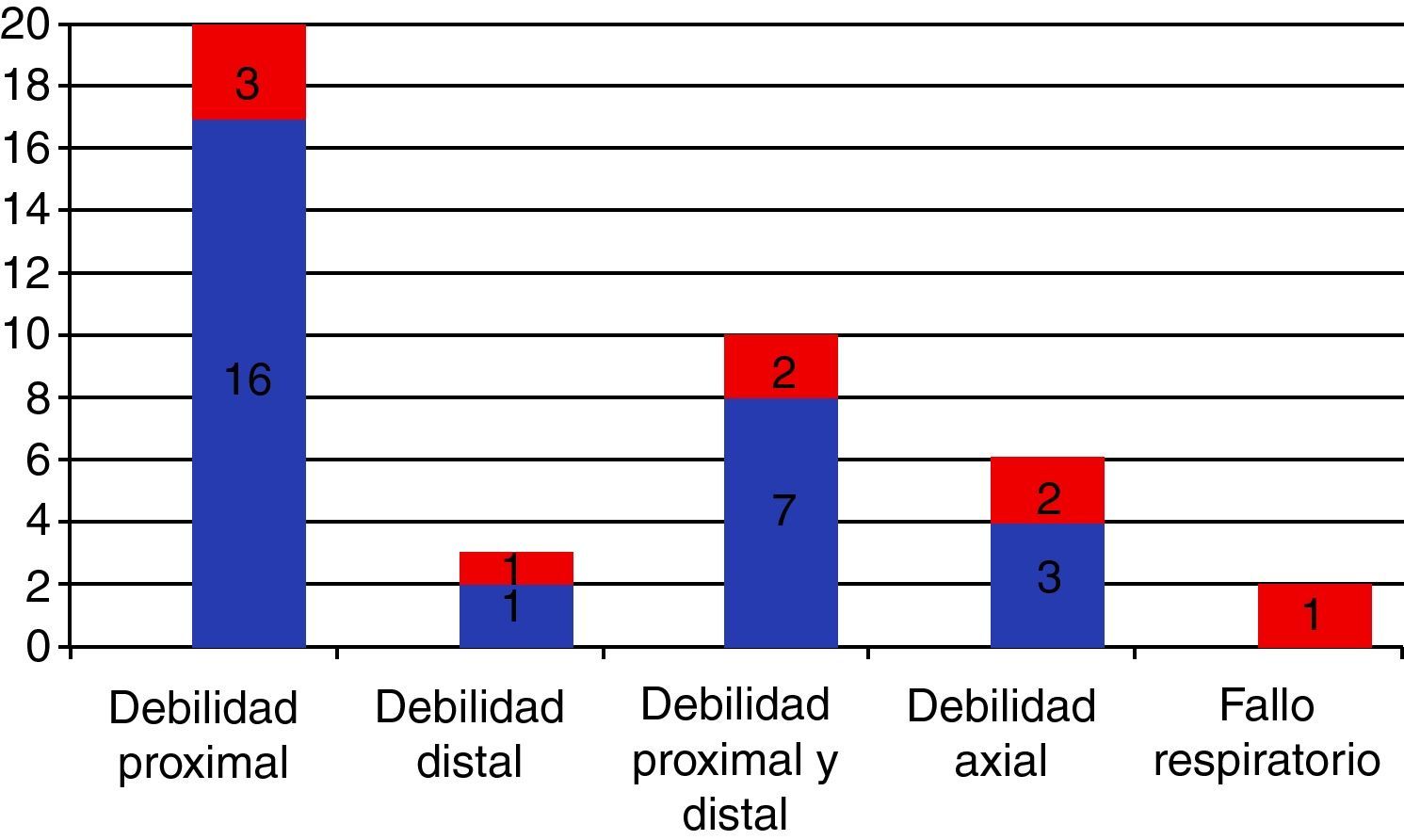
Manifestaciones clínicasLa MCI, a diferencia del resto de MII, tiene predilección por el sexo masculino y la edad avanzada, con una media de edad al inicio de los síntomas de 60 años y un rango que va desde la tercera a la novena década de la vida. Clínicamente se caracteriza por la presencia de debilidad y atrofia muscular de instauración subaguda-crónica, con afectación distal y proximal, a menudo de manera asimétrica. Tiene predilección por la musculatura proximal de los miembros inferiores (cuádriceps) y la distal de los superiores (flexores de los dedos)8,16,54. Los pacientes relatan frecuentemente caídas desde su propia altura, fruto de la ausencia de flexión de la rodilla durante la fase inicial del apoyo de la misma y de una extensión exagerada durante el resto de las fases de apoyo por debilidad del cuádriceps femoral, y dificultad para realizar tareas finas con las manos, como manipular una llave o los botones, por la afectación de los flexores de los dedos. El grado de atrofia muscular suele ir paralelo al grado de debilidad y, sobre todo, a la duración de la clínica. La debilidad puede acompañarse de mialgia hasta en el 40% de los casos, pero suele ser de poca intensidad55. Los músculos paraespinales están también frecuentemente afectados, y la presencia del síndrome de cabeza caída o de camptocormia, resultado de la atrofia selectiva de los mismos, puede ser una forma aislada de presentación. La afectación de la musculatura facial es infrecuente pero posible, excepto el compromiso de los motores oculares. La disfagia ocurre hasta en el 40-60% de los pacientes, y puede constituir también la forma de presentación y preceder a la debilidad incluso en años56,57. Ello es causa de importante morbilidad, con infecciones respiratorias de repetición por broncoaspiración y pérdida de peso. La presencia de dificultad ventilatoria por afectación de la musculatura respiratoria es frecuente durante el curso de la enfermedad, pudiendo ser también la clínica de inicio. La figura 1 recoge las formas de presentación de una serie de 36 pacientes con MCI diagnosticada por nuestro grupo en nuestro centro en un periodo de 10 años58, remarcando la relevancia de la debilidad distal y la disfagia en la sospecha clínica y en el diagnóstico de la enfermedad.
 58.")
Forma de presentación de la miositis con cuerpos de inclusión en 36 pacientes diagnosticados en el Hospital Clínic de Barcelona durante 10 años (en rojo los casos que además tenían disfagia) 58.
Las funciones superiores no se encuentran afectadas, al igual que los reflejos osteotendinosos, si bien estos pueden estar disminuidos por la presencia de atrofia muscular marcada. La hiperreflexia y las fasciculaciones no son propias de la MCI, y su presencia obliga a descartar otros procesos como las enfermedades de la neurona motora59. A diferencia del resto del MII, la participación cardiaca y pulmonar, así como su asociación con el cáncer, es excepcional60,61.
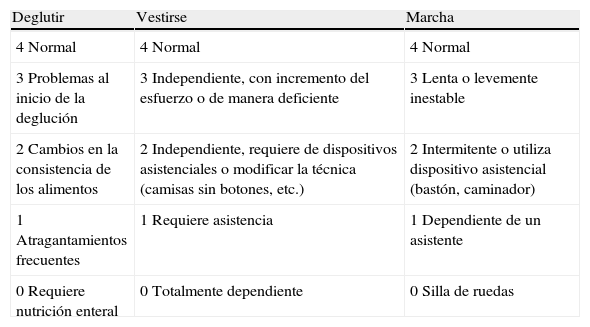
Las escalas de estado funcional específicas, como la desarrollada por Amato et al.8,62, pueden ser de utilidad para la valoración clínica y evolutiva, así como su aplicación en investigación en pacientes con MCI (tabla 2).
Escala de valoración funcional específica de la miositis con cuerpos de inclusión
| Deglutir | Vestirse | Marcha |
| 4 Normal | 4 Normal | 4 Normal |
| 3 Problemas al inicio de la deglución | 3 Independiente, con incremento del esfuerzo o de manera deficiente | 3 Lenta o levemente inestable |
| 2 Cambios en la consistencia de los alimentos | 2 Independiente, requiere de dispositivos asistenciales o modificar la técnica (camisas sin botones, etc.) | 2 Intermitente o utiliza dispositivo asistencial (bastón, caminador) |
| 1 Atragantamientos frecuentes | 1 Requiere asistencia | 1 Dependiente de un asistente |
| 0 Requiere nutrición enteral | 0 Totalmente dependiente | 0 Silla de ruedas |
| Escritura | Higiene | Subir escaleras |
| 4 Normal | 4 Normal | 4 Normal |
| 3 Lenta, todas las letras son legibles | 3 Independiente, aunque con mayor esfuerzo | 3 De forma lenta, vacilante o con mayor esfuerzo |
| 2 No todas las letras son legibles | 2 Independiente, requiere de dispositivos asistenciales (silla de baño, elevar el inodoro) | 2 Requiere sostenerse de la baranda |
| 1 Incapaz de escribir, pero sí de sujetar el bolígrafo | 1 Requiere asistencia ocasional | 1 Requiere de una persona asistente y sostenerse de la baranda |
| 0 Incapaz de sujetar el bolígrafo | 0 Totalmente dependiente | 0 Incapaz |
| Cortar alimentos y manejo de utensilios | Reclinarse en la cama y arroparse |
| 4 Normal | 4 Normal |
| 3 Leve dificultad y lentitud, pero no requiere ayuda | 3 Leve dificultad y lentitud |
| 2 Puede cortar la mayoría de las comidas, requiere cierto grado de ayuda | 2 Moderada dificultad |
| 1 Requiere ayuda para cortar, pero puede alimentarse | 1 Puede iniciar la acción pero requiere de asistencia para finalizar |
| 0 Requiere de asistencia para alimentarse | 0 Totalmente dependiente |
| Motricidad fina (abrir puertas, utilizar llaves, levantar pequeños objetos) | Sedestación y bipedestación |
| 4 Independiente | 4 Independiente (sin utilizar los brazos) |
| 3 Leve dificultad y lentitud en finalizar las tareas | 3 Lo realiza con movimientos alternativos sin utilizar los brazos (inclinarse hacia delante) |
| 2 Independiente, pero requiere de dispositivos asistenciales | 2 Utiliza los brazos |
| 1 Requiere asistencia frecuentemente | 1 Requiere de asistencia |
| 0 Incapaz | 0 Incapaz |
Como ocurre en la mayoría de las enfermedades autoinmunes, es frecuente la asociación con otras entidades, generalmente de origen inmunológico, recogidas en la tabla 316.
Enfermedades inmunológicas asociadas a la MCI
| Inmunodeficiencia común variable |
| Púrpura trombocitopénica idiopática |
| Síndrome de Sjögren |
| Dermatomiositis |
| Lupus eritematoso sistémico |
| Esclerosis sistémica progresiva |
| Artritis reumatoide |
| Sarcoidosis |
De Needham y Mastaglia16.
La presencia de una discreta elevación de enzimas musculares (creatincinasa, aldolasa, lactatodeshidrogenasa, etc.) puede ser la única alteración analítica en pacientes con MCI, hasta un máximo de 10 veces el valor normal, si bien es frecuente que dichas enzimas séricas sean estrictamente normales. Es prácticamente constante la ausencia de reactantes inflamatorios, así como de anticuerpos específicos o asociados a miositis (antisintetasas, anti-Mi2, anti-SRP, anti-PM/Scl, anti-RNP, anti-SSA), pero hasta en el 20% de los casos pueden encontrarse anticuerpos antinucleares8,63,64. Recientemente se ha descrito un autoanticuerpo frente a una proteína muscular de 43kDa (anti-IBM-43) que podría ser específica de MCI65. En una serie de 50 pacientes con miositis (25 con MCI, 10 con PM, 10 con DM y 5 con miastenia gravis) los autores obtuvieron una sensibilidad del 52% y una especificidad del 100% de este autoanticuerpo para el diagnóstico de MCI. Estudios futuros que exploren estos hallazgos ayudarán a determinar su utilidad en el proceso diagnóstico en un paciente con sospecha de MCI. El resto del estudio analítico ha de ir encaminado a descartar otras patologías que puedan tener compromiso muscular (ionograma, hormonas tiroideas, serologías víricas, etc.).
ElectroneuromiografíaLos hallazgos electromiográficos más frecuentes son el aumento de la actividad espontánea en forma de fibrilaciones y descargas repetitivas complejas, la disminución de la amplitud de los potenciales de acción de unidad motora y el reclutamiento precoz. En algunos pacientes pueden detectarse potenciales de acción motora polifásicos de amplitud aumentada que pueden verse también en otras miopatías, inflamatorias o no, así como en neuropatías como la esclerosis lateral amiotrófica, lo que puede ocasionar diagnósticos erróneos66,67. Además, hasta en el 30% de los pacientes pueden detectarse signos de neuropatía, característicamente en forma de afectación axonopática sensitiva8,68.
Resonancia magnética muscularLa RM muscular se ha convertido en una valiosa herramienta a la hora de evaluar un paciente con probable miopatía, sobre todo si se sospecha etiología inflamatoria69, ya que suele aportar información anatómica detallada así como cambios sugestivos de actividad (edema) o cronicidad (atrofía e infiltración grasa). En el caso de la MCI, los signos compatibles con infiltración grasa predominan sobre el edema, con patrón de afectación asimétrico en miembros superiores e inferiores, típico de la enfermedad, y con predilección por los músculos situados ventralmente en cada localización. Un interesante estudio prospectivo publicado recientemente70 realizado en 32 pacientes en los que se exploraron 64 músculos en cada paciente mostró signos patológicos en algún músculo en todos los casos, con una media de 40 músculos afectados por paciente. Los grupos musculares con mayor afectación fueron el gastrocnemio medial y lateral, el vasto lateral, medial e intermedio y el tibial anterior, en orden decreciente, y los menos afectados fueron el extensor carpi ulnaris, el pronador, el infraespinoso, el tríceps y el deltoides. Además, los autores encontraron relación entre el grado de infiltración grasa y la debilidad, el tiempo de evolución de la enfermedad y los niveles séricos de creatincinasa.
Por tanto, la relación entre grasa y edema detectada por RM puede ser útil para diferenciar entre diferentes entidades musculares, y además para sugerir un patrón característico de MCI como el descrito previamente. Ello puede ayudar al diagnóstico en casos de clínica no específica o histología no concluyente70–73. En este sentido, es posible que en un futuro próximo deba incluirse un criterio de RM muscular entre los diagnósticos de MCI.
Anatomía patológica muscularEn la MCI, como en gran parte del resto de miopatías, la biopsia muscular es un estudio imprescindible para el diagnóstico, sobre todo para distinguirla del resto de MII y de las distrofias musculares, entre otras miopatías. Las características principales de la biopsia muscular son las siguientes74:
- •
Infiltrado inflamatorio con predominio de células T, de localización endomisial, con eventual invasión celular19.
- •
Expresión de antígenos de clase I del CHM tanto en fibras morfológicamente normales como en las agredidas por células T74,75.
- •
Vacuolas ribeteadas, más comúnmente visibles en la tinción de Gomori, presentes habitualmente en el 1-6% de las fibras76.
- •
Inclusiones amiloideas citoplasmáticas congofílicas77.
- •
Signos de cronicidad, con fibrosis, fibras atróficas redondeadas y variabilidad en el tamaño de las células.
- •
Fibras rojo-rasgadas, fibras negativas en la reacción citocromo-oxidasa (COX) y positivas en la reacción succinato deshidrogenasa (SDH)74.
- •
Inclusiones tubulofilamentosas intranucleares y citoplasmáticas, visibles solo con microscopia electrónica.
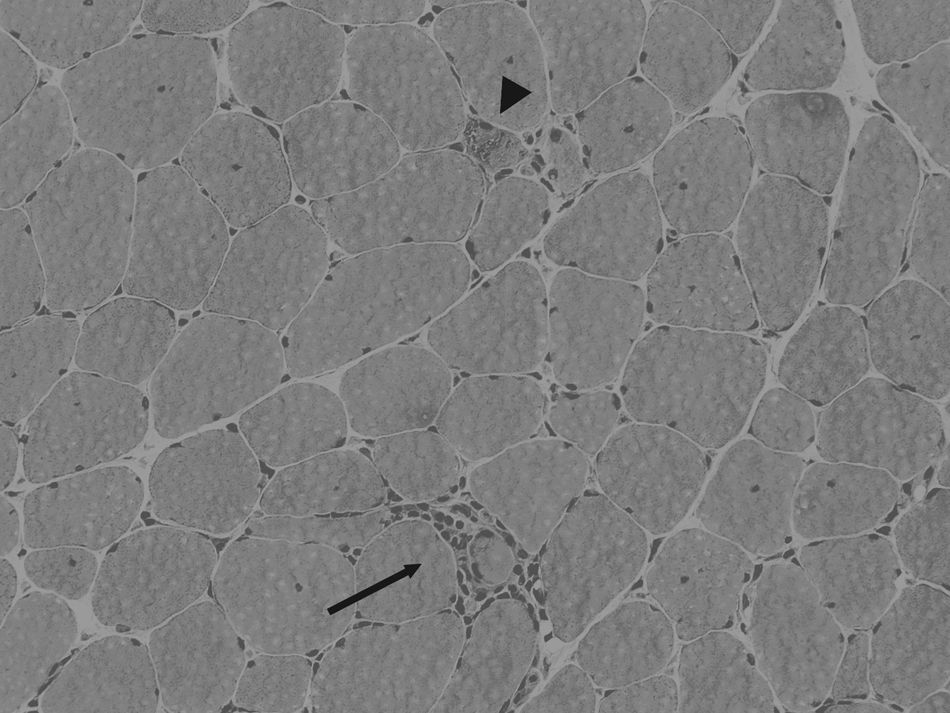
En la figura 2 pueden apreciarse algunos de estos cambios histológicos.
. Se aprecia asimismo variabilidad en el tamaño de las fibras y las típicas vacuolas ribeteadas en una célula muscular (cabeza de flecha). Biopsia muscular en un caso de miositis con cuerpos de inclusión. Hematoxilina-eosina sobre tejido congelado.")
Infiltrado inflamatorio endomisial rodeando una célula muscular (flecha). Se aprecia asimismo variabilidad en el tamaño de las fibras y las típicas vacuolas ribeteadas en una célula muscular (cabeza de flecha). Biopsia muscular en un caso de miositis con cuerpos de inclusión. Hematoxilina-eosina sobre tejido congelado.
La sospecha clínica de la MCI se plantea ante un adulto con debilidad muscular de presentación insidiosa, con compromiso de músculos distales y proximales de forma asimétrica, que frecuentemente relata caídas y disfagia. La media de retraso diagnóstico se cifra entre 5-8 años desde el inicio de la clínica, ya que en muchas ocasiones los síntomas clínicos son poco floridos al inicio de la enfermedad8. En ausencia de marcadores biológicos fiables, el diagnóstico se basa en la histología muscular dentro del contexto clínico descrito, y puede apoyarse, como se ha dicho antes, en alteraciones compatibles en la RM muscular.
Los criterios diagnósticos para la MCI, propuestos por Griggs et al. en 199539 y modificados en 2002 por Tawil y Griggs78 y en 2007 por Dalakas, se recogen en la tabla 4. En la tabla 5 se presentan las categorías diagnósticas29–37.
Criterios diagnósticos de miositis con cuerpos de inclusión
| Clínica |
| Duración de la enfermedad >6 meses |
| Edad de inicio >30 años |
| Debilidad y atrofia muscular lentamente progresiva: temprana afectación de cuádriceps femoral y flexores de los dedos (frecuentemente asimétrica) |
| Disfagia |
| Laboratorio |
| Niveles séricos de CK elevados, pero pueden ser normales |
| EMG: patrón miopático mixto, potenciales unidad motora de corta y larga duración y actividad espontanea |
| Biopsia muscular |
| Necrosis de fibras y regeneración |
| Infiltrado celular mononuclear en endomisio (en grado variable) |
| Invasión de células mononucleares en fibras no necrosadas (mayoritariamente CD8) |
| Expresión de CHM-I en fibras musculares sanas |
| Fibras musculares vacuoladas |
| Inclusiones de ubiquitina y depósitos amiloideos en fibras musculares |
| Inclusiones filamentosas nucleares o citoplasmáticas de 16-20nm en microscopio electrónico |
| Fibras COX-negativas |
Categorías diagnósticas
| Diagnóstico definitivo de miositis por cuerpos de inclusión |
| Características clínicas con biopsia confirmatoria: miopatía inflamatoria con células T y vacuolas, fibras COX-negativas, depósitos amiloideos o inclusiones filamentosas, y expresión de CHM-I. Con estos hallazgos patológicos no es necesario otro dato de laboratorio |
| Presentación atípica de debilidad y atrofia, pero con biopsia característica |
| Probable miositis por cuerpos de inclusión |
| Características clínicas y datos de laboratorio, pero biopsia no característica (p.ej., miopatía inflamatoria necrotizante con infiltrado de células T pero ausencia de vacuolas, depósitos amiloideos, inclusiones filamentosas y fibras COX-negativas) |
| Posible miositis por cuerpos de inclusión |
| Presentación atípica de debilidad con biopsia no característica |
Es importante remarcar que un número no desdeñable de pacientes con MCI previamente habían sido diagnosticados de otra entidad, sobre todo de PM17, dada la atipicidad de la clínica, así como su asimetría en la distribución corporal, hecho este último que puede condicionar la obtención de muestras histológicas subóptimas para un diagnóstico preciso.
Las principales entidades que se deben descartar son:
- •
La PM. Los datos distintivos son el curso agudo-subagudo, la debilidad de predominio proximal y simétrica y los niveles de CK elevados. El estudio histológico acostumbra a ser típico, si bien hay casos en que se superponen algunos datos sugestivos de MCI.
- •
Enfermedad de motoneurona. Los datos distintivos son la hiperreflexia, los calambres y en particular las fasciculaciones. Asimismo, el estudio EMG es muy característico. No es necesario practicar biopsia muscular, salvo en casos especialmente dudosos.
- •
Miopatías vacuolares (MCI hereditaria, miopatías miofrilares). En ellas no existen fenómenos inflamatorios y las células musculares no expresan antígenos de clase I del CHM.
La progresión de la enfermedad es inexorablemente hacia una dependencia funcional total, y la mayoría de los pacientes requieren ayuda para las actividades básicas de la vida diaria a los 15 años de evolución de la misma79. Peng et al.80 evaluaron la progresión de la enfermedad en 78 pacientes, observando que la edad de instauración tenía relación con la evolución. Aquellos pacientes en que la enfermedad se instauraba antes de los 60 años requerían de un caminador en un promedio de 10,2 años, mientras que en aquellos en los que se presentaba después de los 60 años la progresión clínica era mucho más acusada, requiriendo un caminador a los 5,7 años de evolución de la enfermedad.
TratamientoExisten pocos ensayos clínicos sobre terapéutica en la MCI, y la mayoría de las veces con un seguimiento insuficiente, por lo que el tratamiento en esta entidad es fundamentalmente empírico. De estos estudios y, sobre todo, de la experiencia clínica se puede decir que el tratamiento actual de la MCI es infructuoso en la mayoría de los casos16,81. En los pacientes en los que se evidencia respuesta, esta suele ser escasa y subjetiva, con un cierto retraso en la evolución clínica o estabilización temporal. No existen marcadores establecidos como valoración de respuesta a un determinado tratamiento. El único subgrupo de pacientes que presentan una mayor respuesta al tratamiento viene constituido por aquellos con una MCI asociada a otra enfermedad autoinmune sistémica82,83. La MCI presenta una evolución insidiosa que dificulta el diagnóstico, por lo que en la mayoría de los casos este se realiza pasados varios años desde el comienzo de la sintomatología, probablemente cuando se está en la fase de degeneración con importante daño muscular y escasa o nula inflamación, lo que a su vez podría influir en la escasa respuesta al tratamiento.
Las distintas opciones terapéuticas ensayadas en la MCI son:
CorticosteroidesComo en la gran mayoría de las enfermedades autoinmunes, constituyen el primer eslabón terapéutico. Su uso se basa es estudios retrospectivos no controlados y muestran una respuesta más bien escasa, que en algunas series puede alcanzar el 40-58% de los pacientes si esta se basa en criterios subjetivos84,85. En los casos en que se constata un respuesta objetiva, esta es en forma de mejoría leve o estabilización transitoria16,86,87. Los niveles de CK pueden descender, aunque este descenso no necesariamente se traduce en una mejoría clínica85. Como ya se ha comentado, en los pacientes en los que la MCI coexiste con otra enfermedad autoinmune sistémica, la tasa de respuesta al tratamiento esteroideo es mayor55,88,89.
InmunosupresoresEl metotrexato y la azatioprina, solos o en combinación, con mayor experiencia en el primero, han mostrado un efecto similar en forma de respuestas incompletas no mantenidas solo en algunos casos55,84,85,87,90. Al igual que el grupo farmacológico descrito en el párrafo anterior, pueden condicionar una disminución de los niveles de CK sin beneficio clínico85,90.
Con otros fármacos, como la ciclofosfamida, el micofenolato, el tacrolimus y el clorambucilo, la experiencia es muy escasa y con nula o mínima respuesta91–94.
Inmunoglobulinas intravenosasLa eficacia de las inmunoglobulinas intravenosas ha sido evaluada en 3 estudios doble ciego y dos pequeñas series abiertas95–99, con resultados más bien desalentadores:
- •
En el primer estudio no controlado98, tras 2 meses de tratamiento con dosis altas de inmunoglobulinas intravenosas se observó aumento en la fuerza de músculos proximales en 3 de 4 pacientes en un periodo de 2 a 4 meses).
- •
En otro estudio abierto no controlado99 que incluía 9 pacientes tratados con inmunoglobulinas intravenosas no se demostró mejoría en la fuerza ni en la función muscular, y además con el inconveniente de notables efectos secundarios.
- •
Un estudio doble ciego aleatorizado y controlado con placebo incluyó 19 pacientes con MCI a los que se administró 2g/kg de inmunoglobulinas intravenosas o placebo por 3 meses. No se encontraron diferencias significativas con respecto a mejoría de la fuerza muscular95.
- •
Otro estudio doble ciego aleatorizado y controlado con placebo incluyó 36 pacientes asignados a recibir inmunoglobulinas intravenosas o placebo (durante 3 meses), administrando dosis altas de prednisona durante 3 meses antes de realizar las perfusiones. Tras 4 meses de observación no se demostraron diferencias significativas con respecto a mejoría de la fuerza muscular entre los dos grupos. Tras analizar las biopsias musculares se comprobó cierta disminución del número de fibras necróticas en los pacientes que recibieron inmunoglobulinas intravenosas, aunque sin correlación clínica. Los autores concluyeron que la combinación de inmunoglobulinas intravenosas con prednisona durante un periodo de 3 meses no era un tratamiento efectivo en pacientes con MCI96.
- •
Un último estudio doble ciego aleatorizado y controlado con placebo incluyó 22 pacientes con MCI, asignados a recibir 2g/kg de inmunoglobulinas intravenosas o placebo durante 6 meses. En el grupo de inmunoglobulinas intravenosas se constató estabilización de la enfermedad en 18 de 22 pacientes, sin que existieran diferencias significativas con respecto a mejoría en la fuerza muscular97.
En vista de estos resultados, dado su elevado coste económico y su limitado beneficio no se recomienda su administración, salvo en pacientes con disfagia grave, en los que, tras 6-8 meses de tratamiento, se puede recuperar la función deglutoria100.
Agentes biológicosLa inmunoglobulina específica anti-linfocito T, el interferón beta 1α, el etanercept y el alemtuzumab se han utilizado como agentes biológicos en el tratamiento de la MCI101–104. Solo el último101, en un pequeño estudio no controlado de 13 pacientes mostró mejoría de la fuerza y disminución en el ritmo evolutivo de la enfermedad, mostrándose como un prometedor tratamiento cuya eficacia debe contrastarse, aunque deben tenerse en cuenta los eventuales efectos secundarios de los agentes biológicos.
Terapia sintomáticaUna alternativa a la administración de inmunoglobulinas intravenosas en los pacientes con disfagia grave es la miotomía cricofaríngea105. También se ha evaluado un programa de entrenamiento físico tipo aeróbico, encaminado a aumentar la fuerza y mejorar la función muscular106,107.
Otras terapiasEn la actualidad se han producido nuevos avances que permiten comprender algo mejor el mecanismo fisiopatológico de la MCI, lo que justifica innovadoras líneas de investigación relacionadas con terapias para acelerar la regeneración y enlentecer la degeneración muscular. Con respecto a las estrategias para acelerar el crecimiento muscular, se desestima el uso de andrógenos por sus reacciones adversas, aunque la oxandrolona ha demostrado cierta eficacia y ser bien tolerada en un pequeño estudio doble ciego controlado con placebo en pacientes con MCI108. Otro grupo farmacológico que se encuentra en investigación es el de los inhibidores de la miostatina, potente supresor del crecimiento muscular109. Otra manera de estimular el crecimiento muscular podría ser la implantación de células madre pluripotenciales110.
En relación con las estrategias para disminuir la degeneración muscular, se encuentra en estudio el arimoclomol, inductor de proteínas heat-shock que previenen la agregación de proteínas dentro de la célula muscular. Otra línea de investigación se fundamenta en el hecho de que el mecanismo de autofagia es defectuoso en MCI, por lo que podrían utilizarse fármacos que estimulen la fagocitosis como el litio, que también inhibe la fosforilación de la proteína tau111. La rapamicina, fármaco antiinflamatorio e inductor de autofagia, ha demostrado eliminar agregados de TDP-43, generando la hipótesis de que al acelerar el mecanismo autofágico se podrían evitar los efectos tóxicos producidos por TDP-43112. El resveratrol es otro fármaco que produciría beneficios a pacientes con MCI, ya que favorece e incrementa el mecanismo autofágico113.
Se ha sugerido que las estatinas podrían beneficiar a pacientes con MCI, debido a su efecto pleotrópico en autoinmunidad. En un estudio abierto que incluyó 14 pacientes a quienes se les administró 40mg de simvastatina durante 12 meses, se comprobó que ningún paciente presentó mejora clínica114.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.