


ETIOLOGÍA
La artritis reumatoide (AR) es una enfermedad inflamatoria crónica que afecta fundamentalmente a la sinovia, lo que lleva a la destrucción del cartílago articular. La AR afecta aproximadamente al 1-3% de los individuos caucásicos, y es una de las enfermedades autoinmunes más prevalentes en estas poblaciones. Su prevalencia es de 3 a 5 veces mayor en mujeres que en varones, y su incidencia es mayor entre el segundo y el cuarto decenio de vida, aunque puede aparecer a cualquier edad.
La AR es una enfermedad compleja de etiología desconocida. A partir de estudios epidemiológicos y genéticos se postula que la AR se produce en individuos genéticamente predispuestos por la acción de un factor ambiental. El factor ambiental que desencadena la AR es desconocido, pero como en otras enfermedades autoinmunes, se cree que el responsable es una infección. Estudios realizados en gemelos monocigóticos y dicigóticos indican que el componente genético es responsable aproximadamente del 60% de la susceptibilidad a la enfermedad1. Sin embargo, como en otras enfermedades autoinmunes, su herencia no sigue un patrón mendeliano clásico, sino que son enfermedades poligénicas en las que se requiere la interacción de numerosos genes que confieren el riesgo de padecer la enfermedad. Hay diversos genes que incrementan la probabilidad de padecer la enfermedad, pero ninguno de ellos por sí solo determina si un individuo desarrollará o no AR. Con frecuencia estos genes muestran interacciones complejas, con penetrancia baja e incompleta y patrones de herencia no mendelianos. Como resultado, hay una considerable heterogeneidad genética, y por tanto también clínica, en los pacientes con AR.
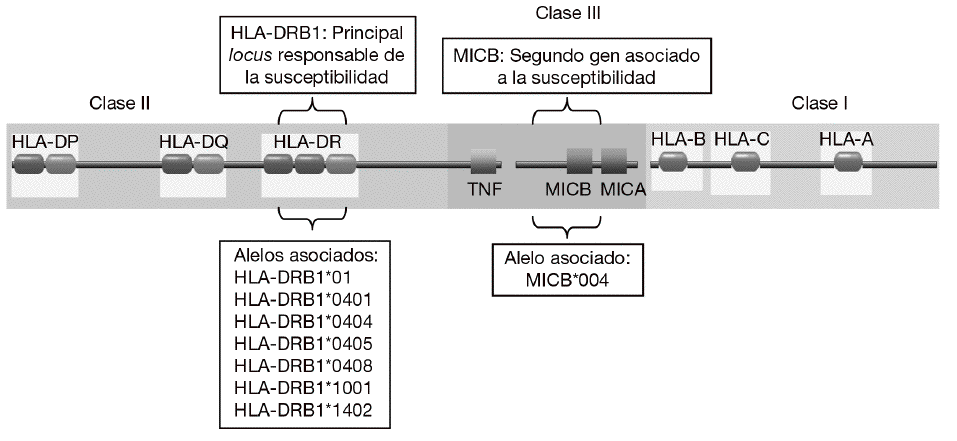
En consonancia con un patrón de herencia poligénica, se han descrito numerosos loci asociados a la susceptibilidad a la AR que se encuentran distribuidos a lo largo de todo el genoma humano2,3. Sin embargo, la mayoría de los estudios han demostrado de forma consistente que la región del complejo mayor de histocompatibilidad (MHC) es la principal responsable de la susceptibilidad genética a la AR (fig. 1). Mediante estudios serológicos se demostró, ya hace muchos años, la asociación del HLA-DR4 y el HLA-DR1 con la AR4. Más recientemente, mediante análisis realizados con técnicas de biología molecular se observó que sólo algunos alelos de HLA-DR4 y HLA-DR1 se asociaban con la susceptibilidad y la gravedad de la AR. Estos alelos eran el alelo HLA-DRB1*0101 (serológicamente, HLA-DR1) y los alelos HLA-DRB1*0404 y DRB1*0405 (serológicamente, HLA-DR4)5-7. Es significativo que diferentes alelos de HLA-DR estén asociados al desarrollo de la enfermedad. Esto se debe, probablemente, a que aunque estos alelos son diferentes, comparten un epítopo de 5 aminoácidos rico en residuos básicos (QKRAA o QRRAA) en la cadena b de la molécula HLA-DR8-10. Esto sugiere que probablemente todos ellos conserven la capacidad de presentar el mismo o los mismos péptidos derivados del mismo autoantígeno presente en las articulaciones. Sin embargo, no todos los individuos con estos alelos de HLA-DR padecen la enfermedad es necesario la interacción con otros genes y con un factor ambiental, ni todos los pacientes tienen estas secuencias de suscepti-bilidad. Esto sugiere que es posible que otras mo-léculas de HLA-DR puedan presentar el mismo péptido o que existan diferentes autoantígenos implicados en la patogenia de la enfermedad.
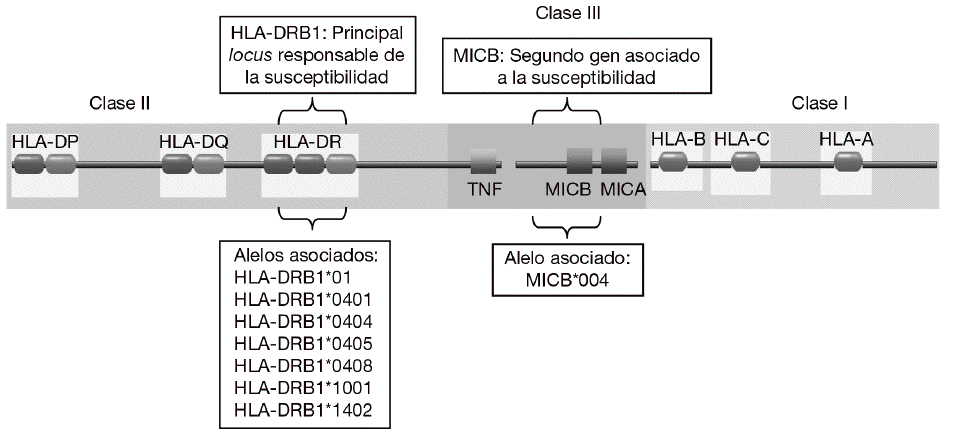
Figura 1 Genes de susceptibilidad presentes en la región del complejo mayor de histocompatibilidad (MHC) asociados con la artritis reumatoide. El gen HLA-DRB1 es el principal responsable de la susceptibilidad a la enfermedad. Diversos alelos que comparten un epítopo de 5 aminoácidos rico en residuos básicos (QKRAA o QRRAA) en la cadena b del HLA-DR se asocian con su susceptibilidad. Adicionalmente, se ha descrito que el gen MICB situado en la región del MHC de clase III está también asociado.
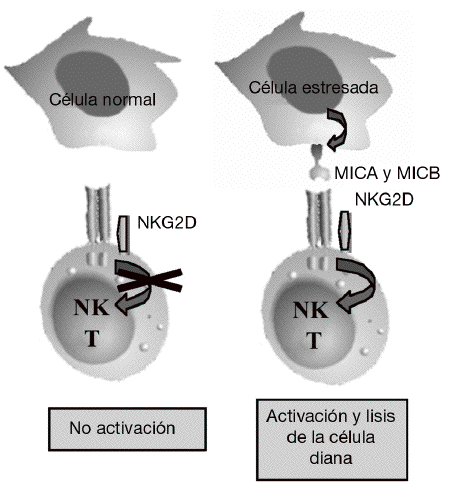
Aunque la asociación de HLA-DR es bien conocida, a partir de estudios genéticos y epidemiológicos se ha sugerido la existencia de genes adicionales al HLA-DR que pueden incrementar o modificar la susceptibilidad al desarrollo de la enfermedad11-17. Los genes candidatos se localizan tanto en la región del MHC como fuera de ella. La región del MHC es una pequeña región del genoma de 3,6 megabases localizada en el brazo corto del cromosoma 6 (fig. 1). Esta región codifica para las moléculas tanto de HLA de clase I como de clase II, pero también para muchas otras moléculas que tienen funciones inmunológicas y no inmunológicas. Mediante estudios de asociación realizados recientemente en 18 microsatélites distribuidos a lo largo de todo la región del MHC se demostró que, como era de esperar, el gen HLA-DRB1 constituye la asociación más importante a la susceptibilidad a AR17. Sin embargo, no es la única, ya que estos autores describieron la presencia de un segundo gen de susceptibilidad localizado en un pequeño intervalo de 70 kb en la región de MHC de clase III, situado en la parte telomérica al gen de factor de necrosis tumoral alfa (TNF a). En esta pequeña región de susceptibili-dad se localizan 4 genes, denominados IkBL, ATP6G, BAT1 y MICB. Estos resultados excluían la posibilidad de una asociación del gen del TNF a con la susceptibilidad a la AR. Entre estos genes candidatos es especialmente importante destacar el posible papel que puede desempeñar MICB en el desarrollo de la AR. MICA y MICB (MICA/B) forman una nueva familia de moléculas de HLA de clase I denominadas no clásicas18. Estas moléculas no presentan péptidos y no se expresan en la mayoría de las células en condiciones normales, pero su expresión se incrementa notablemente en respuesta al estrés biológico (p. ej., en respuesta a una infección o transformación tumoral), y entonces son reconocidas por las células natural killer (NK) y los linfocitos T a través de un receptor activador que se expresa en estas células denominado NKG2D19 (fig. 2). Este reconocimiento permite al sistema inmune eliminar las células que padecen estrés biológico y, por tanto, constituye un mecanismo de inmunovigilancia. La mediación de MICA/B en la eliminación de células estresadas implica que estas moléculas deben estar estrictamente reguladas, ya que su expresión exagerada (p. ej., en los sinoviocitos) puede llevar a la destrucción de las propias células y, consecuentemente, a la generación de una respuesta autoinmune, como la que se observa en la AR.
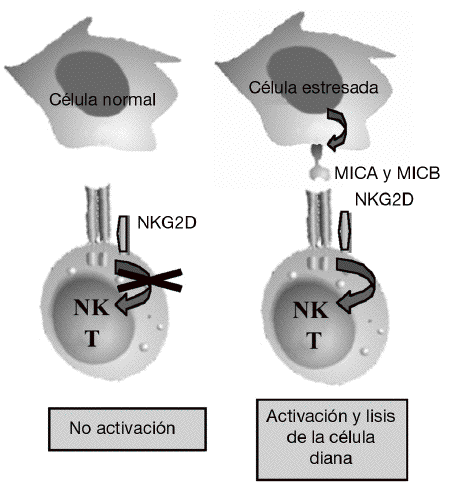
Figura 2 Función de MICA y MICB. Estas moléculas no se expresan en células normales, pero su expresión se induce en células infectadas, transformadas o estresadas, lo que permite su eliminación por el sistema inmune mediante su interacción con el receptor activador NKG2D presente en linfocitos T y células natural killer (NK).
PATOGENIA
La AR se origina por una respuesta autoinmune contra un antígeno propio localizado en las articulaciones. Las enfermedades autoinmunes se producen por una respuesta inmune adaptativa es decir, mediada por linfocitos B o T contra nuestros propios antígenos. En esencia, el sistema inmune confunde lo propio y lo extraño, por lo que los mecanismos inmunológicos que se generan son los mismos que se producen contra los patógenos. Sin embargo, a diferencia de éstos, las respuestas autoinmunes son generalmente incapaces de acabar completamente con los antígenos propios, que se regeneran constantemente, por lo que dan lugar a reacciones inflamatorias crónicas que duran toda la vida.
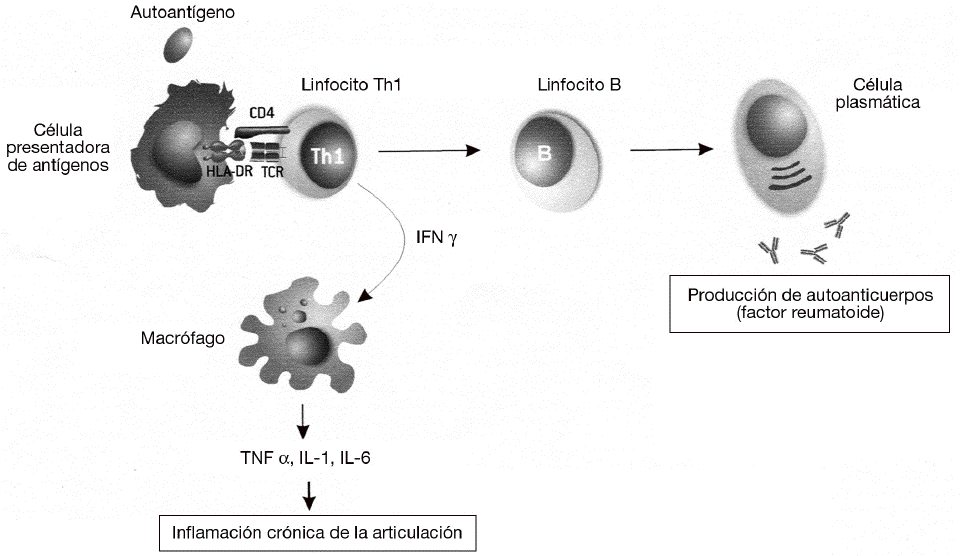
Las enfermedades autoinmunes se pueden clasificar según el mecanismo inmunológico que se genera contra estos antígenos. La mayoría de las enfermedades autoinmunes se producen por una respuesta humoral, mediada por la producción de anticuerpos contra antígenos propios. Sin embargo, algunas enfermedades autoinmunes muy prevalentes la diabetes mellitus insulinodependiente, la esclerosis múltiple o la AR se producen por respuestas celulares mediadas por linfocitos T. En particular, la respuesta autoinmune en la AR está mediada por un tipo de linfocitos CD4 o colaboradores denominados linfocitos Th1 o inflamatorios (fig. 3). Estos linfocitos T reconocen específicamente un antígeno propio, o autoantígeno, que no se ha identificado todavía pero que está presente en las articulaciones. El reconocimiento de este autoantígeno por los linfocitos Th1 desencadena la liberación de citocinas como el interferón g (IFN g), que activan a los macrófagos para favorecer la destrucción del autoantígeno. La activación de los macrófagos por los linfocitos Th1 incrementa significativamente su capacidad fagocítica, pero también induce la liberación de citocinas proinflamatorias, como el TNF a, por parte de dichos macrófagos que causan una respuesta inflamatoria crónica. Esta respuesta se caracteriza por la tumoración de la articulación y la acumulación de más macrófagos y polimorfonucleares que dañan de forma crónica el cartílago, lo que lleva a la destrucción de la articulación. La AR es una enfermedad compleja en la que frecuentemente se involucra también la producción de autoanticuerpos. A menudo se produce un anticuerpo IgM anti-IgG llamado factor reumatoide. En esta enfermedad como ocurre con el lupus eritematoso sistémico, la unión del factor reumatoide a la IgG causa la formación de complejos antígeno-anticuerpo o inmunocomplejos. Si se produce una gran cantidad de estos inmunocomplejos, se depositan en los tejidos y causan daño tisular sistémico por la activación de los mecanismos inflamatorios en los lugares de depósito.
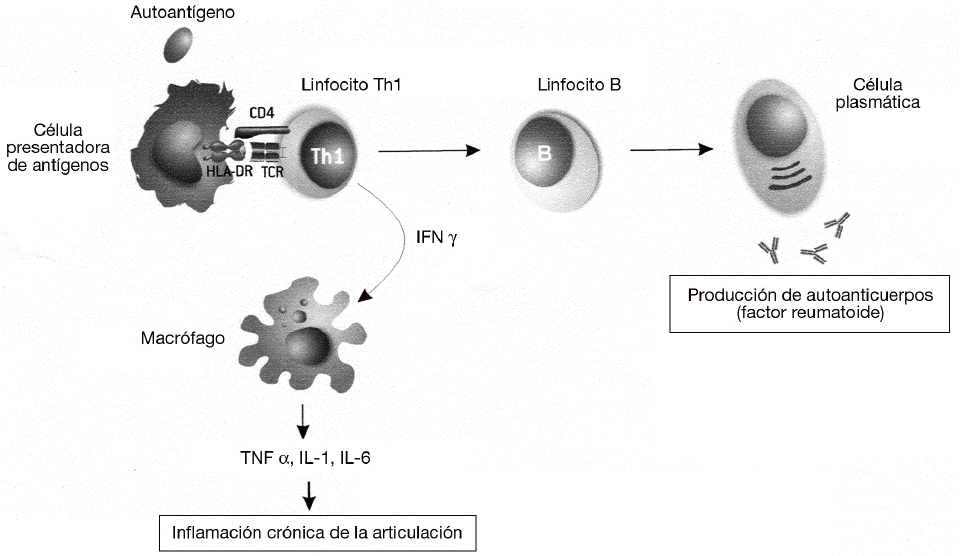
Figura 3 Patogenia de la artritis reumatoide. Las células presentadoras activan linfocitos Th1 autorreactivos mediante la presentación de autoantígenos derivados de la articulación mediante las moléculas del complejo mayor de histocompatibilidad (MHC) de clase II (HLA-DR). Mediante la secreción de interferón g (IFN g), los linfocitos Th1 inducen la activación de los macrófagos, que comienzan a sintetizar citocinas proinflamatorias como el factor de necrosis tumoral alfa (TNF a), interleucina (IL) 1 e IL-6. Esta respuesta es exagerada (debido a la susceptibilidad genética y a los factores ambientales) y produce una inflamación crónica que acaba destruyendo las articulaciones. Además, también es muy frecuente la activación de linfocitos B autorreactivos y la producción de autoanticuerpos. A menudo se produce un anticuerpo IgM anti-IgG llamado "factor reumatoide", que puede dar lugar a la formación de inmunocomplejos y causar daño sistémico por activación de los mecanismos inflamatorios en los lugares de depósito.
MICA favorece la eliminación por el sistema inmune de células estresadas o dañadas
NKG2D es un receptor activador que se expresa de forma constitutiva en la superficie de células citotóxicas del sistema inmune tanto innato (células NK y linfocitos Tgd) como del sistema inmune adaptativo (linfocitos Tab CD8 o citotóxicos)19. Mediante este receptor, las células citotóxicas del sistema inmune son capaces de reconocer y eliminar células que han experimentado algún tipo de estrés biológico, como pueden ser la infección por un virus o la transformación tumoral. Este reconocimiento se produce porque NKG2D se une a una serie de ligandos que no se expresan en células sanas, pero cuya expresión se incrementa en la superficie de las células dañadas como consecuencia de dichos procesos de estrés (fig. 2). La unión de este receptor con sus ligandos se traduce en la activación de las células citotóxicas que provoca la liberación dirigida de gránulos citolíticos sobre la superficie de la célula estresada, lo que causa la lisis o apoptosis de las células diana. El sistema constituido por el receptor NKG2D y sus ligandos supone un nuevo paradigma de reconocimiento del sistema inmune en el que nuestro sistema inmune no reconoce directamente antígenos extraños presentes en los patógenos, sino que reconoce los cambios o daños que los patógenos en particular, y el estrés biológico en general, producen en las propias células.
En humanos, NKG2D tiene como ligandos 2 familias de moléculas similares a las moléculas de HLA de clase I denominadas MIC (MHC class I related genes) que incluyen a MICA/B, y la familia de las ULBP (UL16 Binding Proteins)19. MICA/B fueron los primeros ligandos de NKG2D identificados. Los genes que codifican estas proteínas se sitúan cerca de la región del MHC de clase I (fig. 1). MICA/B son moléculas de MHC de clase I denominadas no clásicas, ya que tienen una estructura formada por 3 dominios denominados a1, a2 y a3, similares a los que tienen las moléculas de MHC de clase I, pero a diferencia de éstas, no se unen a b2-microglobulina ni presentan péptidos intracelulares. MICA/B se expresan de una forma restringida en tejidos sanos, pero los valores de estas proteínas se incrementan en la superficie de células tumorales o sometidas a otros tipos de estrés (p. ej., por una infección por citomegalovirus humano, el estrés oxidativo o el estrés genotóxico).
No obstante, se han descrito numerosos mecanismos de evasión de la respuesta inmune frente a estas células bajo condiciones de estrés. Por ejemplo, en las células tumorales MICA es digerido de la superficie de las células por la acción de una proteasa, lo que da lugar a la formación de moléculas solubles de MICA. La digestión de MICA de la superficie de la célula dificulta el reconocimiento de las células tumorales por el sistema inmune, y además, la forma soluble de MICA es un potente inmunosupresor que inhibe la expresión de NKG2D en la superficie de los linfocitos T y de las células NK20,21.
MICA/B son 2 moléculas extremadamente polimórficas19. No se conoce el papel que la mayoría de estos polimorfismos ejercen sobre la función de estas moléculas, pero en algunos casos se ha descrito que estos polimorfismos afectan a la expresión de MICA/B o a su capacidad de activación de las células citotóxicas del sistema inmune. El sistema inmune siempre tiene que mantener un delica do equilibrio entre destruir lo extraño o dañino y respetar lo propio. Es tentador pensar que los polimorfismos que aumenten la expresión o la capacidad de activación de MICA o de MICB sean responsables de una respuesta inmune más intensa, lo que probablemente sería beneficioso para eliminar más eficazmente las células infectadas por patógenos o que han experimentado una transformación tumoral, pero una respuesta más intensa podría favorecer el desarrollo de respuestas autoinmunes. En consonancia con esta idea se han descrito numerosos polimorfismos de MICA/B que se asocian con un incremento en el riesgo de desarrollo de determinadas patologías autoinmunes como la diabetes mellitus insulinodependiente, la enfermedad celíaca o la AR22.
La expresión exagerada de MICA puede causar autoinmunidad
Las enfermedades autoinmunes se deben a una respuesta inmune adaptativa contra los propios antígenos. Las respuestas inmunes adaptativas comienzan por la activación de linfocitos T contra antígenos específicos de su receptor (TCR), y se piensa que las enfermedades autoinmunes se inician de la misma forma. La mayoría de las enfermedades autoinmunes están mediadas por una respuesta humoral contra antígenos propios. Sin embargo, algunas de estas patologías están mediadas por una respuesta celular caracterizada por la presencia de linfocitos Th1 o citotóxicos específicos contra antígenos propios.
MICA/B son moléculas inducidas por el estrés biológico. Su función es alertar al sistema inmune del posible daño de alguna de nuestras células. Ya que el receptor NKG2D se expresa de una forma constitutiva en los linfocitos T citotóxicos y en las células NK, la expresión de MICA/B puede llevar directamente a la destrucción de la célula dañada19. Esto sugiere que la expresión inespecífica de MICA/B durante un proceso inflamatorio o la expresión inadecuada en individuos genéticamente predispuestos pueden desencadenar o exacerbar las enfermedades autoinmunes. De acuerdo con esta hipótesis se han aportado numerosas pruebas que indican que MICA/B pueden estar implicados en la patogenia de diversas enfermedades autoinmunes y de hipersensibilidad mediadas por linfocitos T, como la enfermedad celíaca, la diabetes mellitus insulinodependiente, la esclerosis múltiple o la AR22.
La enfermedad celíaca se produce por una reacción de alergia o hipersensibilidad causada por linfocitos T efectores específicos del gluten, en particular de una fracción denominada gliadina, y ello causa atrofia de las vellosidades intestinales y un síndrome de malabsorción. En la patogenia de la enfermedad celíaca participan tanto los linfocitos T colaboradores (CD4) como los citotóxicos (CD8)23,24. Hay sólidas pruebas de que el desencadenamiento de la enfermedad celíaca se produce por el reconocimiento por linfocitos Th1 (CD4) de péptidos derivados del gluten presentados a través de las moléculas HLA-DQ2 o HLA-DQ8. No obstante, los linfocitos CD8 también desempeñan un papel importante en el desarrollo de la enfermedad. Existe una infiltración masiva de la mucosa de los pacientes celíacos de linfocitos intraepiteliales. Estos linfocitos intraepiteliales son linfocitos T CD8 que expresan de una forma constitutiva el receptor NKG2D. Se ha demostrado recientemente que MICA y MICB se expresan en la superficie de las células epiteliales intestinales, mientras que presentan una localización intracelular en los enterocitos normales. Es probable que la toxicidad de la propia gliadina sea la responsable de la expresión de MICA/B, ya que ésta cesa con la dieta sin gluten. La presencia de MICA/B desencadena la destrucción de las células epiteliales de una forma independiente del antígeno, lo que desempeña un papel esencial en la lesión tisular que se observa en la enfermedad.
También se ha demostrado recientemente que NKG2D participa en el desarrollo de la diabetes mellitus insulinodependiente en modelos murinos de la enfermedad, como son los ratones NOD (non obese diabetic mice)25,26. Los ligandos murinos de NKG2D se expresan en las células b del páncreas de ratones prediabéticos y diabéticos, pero no en ratones normales. El páncreas de estos ratones NOD está densamente infiltrado por linfocitos T citotóxicos autorreactivos que expresan NKG2D, que son responsables de la destrucción de las células b productoras de insulina. Es muy significativo que el tratamiento de estos ratones NOD con un anticuerpo capaz de bloquear NKG2D previene el desarrollo de la enfermedad, lo que demuestra el papel crucial de este receptor en la patogenia de la enfermedad. En humanos, el papel de MICA en la patogenia de la diabetes no ha sido aún estudiado, pero esta molécula se ha asociado con su susceptibilidad genética, lo que sugiere un mecanismo patogénico similar22. Igualmente se ha sugerido que MICA y MICB pueden estar involucrados en la patogenia de otras enfermedades autoinmunes, como la esclerosis múltiple27.
Implicación de MICA en el desarrollo de la artritis reumatoide
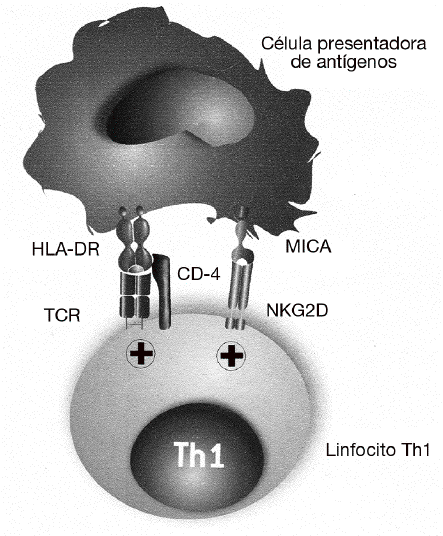
El papel de MICA y NKG2D en el desarrollo de las enfermedades autoinmunes fue descrito por primera vez en la AR28. Como ya se ha dicho, se sabe desde hace tiempo que la gravedad de la AR se correlaciona con la presencia de un gran número de linfocitos Th1 autorreactivos con el fenotipo CD4+CD25, que están presentes tanto en la sangre periférica como infiltrando la sinovia. En estos linfocitos CD4 se describió por primera vez la expresión del receptor NKG2D, que en condiciones normales se expresa exclusivamente en las células citotóxicas y que normalmente está ausente en los linfocitos CD4 de individuos sanos (fig. 4). En estos linfocitos, NKG2D no induce directamente la citotoxicidad de las células diana, como ocurre con los linfocitos T CD8 o células NK, sino que tiene un papel coestimulador. Su función consiste en favorecer la activación de los linfocitos Th1 que han sido previamente estimulados a través de su receptor específico, incrementando la producción de citocinas (como el IFN g) y aumentando su capacidad de activación de los macrófagos. En este caso, las células implicadas en la patogenia de la enfermedad no son los linfocitos T CD8, excluyendo un mecanismo similar a la diabetes mellitus insulinodependiente o a la enfermedad celíaca.
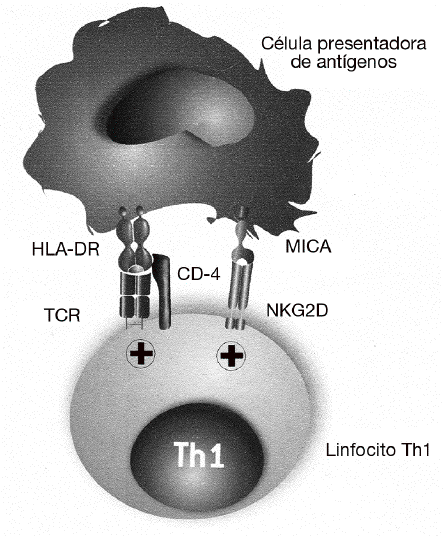
Figura 4 MICA y MICB favorecen la activación de los linfocitos Th1 exacerbando o incrementando la reacción inflamatoria en la artritis reumatoide. En esta enfermedad, MICA y MICB no son responsables directamente de la destrucción de los sinoviocitos, sino que tienen una función coestimuladora de los linfocitos Th1 autorreactivos. Favorecen la activación de los linfocitos T que han sido previamente estimulados por su receptor específico (TCR) por autoantígenos presentados por las moléculas del MHC de clase II (HLA-DR). La consecuencia de esta estimulación es el incremento de la producción de citocinas (interferón g) y de su capacidad de activación de los macrófagos.
La expresión de NKG2D se induce en los linfocitos CD4 en la AR por la acción de dos citocinas: TNF a e interleucina 15 (IL-15), que abundan en la sinovia y el suero de los pacientes. Además, los sinoviocitos de los pacientes con AR expresan de una forma exagerada MICA, MICB y otros ligandos de NKG2D, comos son las ULBP1-3, los cuales estimulan a los linfocitos T CD4 autorreactivos a través de NKG2D y exacerban o perpetúan el desarrollo de la AR. El suero de los pacientes con esta enfermedad contiene cantidades sustanciales de la forma soluble de MICA que es liberada de la superficie de los sinoviocitos por acción de una proteasa de una forma muy similar a lo que ocurre en múltiples cánceres. Como ya se ha dicho, es bien conocido que la producción de la forma soluble de MICA por las células cancerígenas tiene un potente efecto inmunosupresor, ya que inhibe la expresión de NKG2D en la superficie de los linfocitos T y de las células NK. Sin embargo, en el caso de la AR el efecto de MICA soluble es contrarrestado por una actividad opuesta del TNF a y el IFN g, muy abundantes en la sinovia y el suero en los pacientes con AR, que inducen la expresión de NKG2D. La expresión de MICA/B en los sinoviocitos causa la estimulación de los linfocitos Th1 autorreactivos, promoviendo así la autoperpetuación de la respuesta autoinmune en la AR. Significativamente, se ha observado que la frecuencia de linfocitos T CD4+C28 se incrementa de una forma significativa con el envejecimiento. Se ha postulado que este incremento puede ser un mecanismo que favorece el desarrollo de enfermedades autoinmunes, y en particular de la AR, con el aumento de la edad.
De acuerdo con el papel de MICA/B en la patogenia de la AR, hemos descrito recientemente la asociación de MICB con la susceptibilidad al desarrollo de la enfermedad29. En particular, hemos encontrado un alelo de MICB denominado MICB*004 asociado con su susceptibilidad. En nuestra población, MICB*004 se asocia a la AR independientemente de HLA-DR, aunque MICB*004 también está en desequilibrio de ligamiento con los alelos de susceptibilidad HLA-DRB1*0404, HLA-DRB1*0405 y HLA-DRB1*1402, clásicamente asociados a la susceptibilidad de la AR8-10. Esto sugiere que la presencia o ausencia de MICB*004 en sus haplotipos puede modificar el riesgo conferido por HLA-DR. Nuestros resultados podrían explicar por qué los alelos HLA-DRB1*0401 y HLA-DRB1*0404, que comparten el mismo epítopo de susceptibilidad, difieren significativamente en su grado de asociación con la AR16. Es tentador especular que el ligamiento de MICB*004 con HLA-DRB1*0404 puede ser responsable de que HLA-DRB1*0404 confiera mayor riesgo de padecer la AR que HLA-DRB1*0401.
A pesar de que MICA y MICB son dos moléculas altamente polimórficas, las características funcionales de la mayoría de sus polimorfismos son desconocidas. Sin embargo, la mayormas, por lo que hay que asumir que pueden ser funcionales. Hemos descrito recientemente que el promotor de MICB es polimórfico. Algunos alelos de MICB presentan en su región promotora una deleción de 2 pares de bases en la posición 66 (AG/--) que disminuyen muy significativamente la transcripción de MICB (hasta unas 18 veces)30. Estos polimorfismos son muy frecuentes en nuestra población, lo que implica que existen grandes variaciones en la expresión de MICB entre los diferentes individuos que la componen. Esto podría llevar a suponer que los individuos que producen grandes cantidades de MICB (como los que son MICB*004) expresen mayor cantidad de MICB en la sinovia y produzcan una reacción autoinmune más intensa. Esto explicaría por qué estos individuos se asocian con un mayor riesgo de padecer AR y otras enfermedades inmunológicas, como la esclerosis múltiple o la enfermedad celíaca31. Se puede predecir que, por el contrario, estos individuos estarían más protegidos del desarrollo del cáncer, aunque esto no se ha demostrado aún experimentalmente.
En conjunto, todos estos datos indican que NKG2D y MICA/B desempeñan un papel crucial en la defensa del organismo contra las infecciones y el cáncer. Si este mecanismo inmunológico de inmunovigilancia no se regula correctamente, se puede producir una estimulación de los linfocitos T autorreactivos y causar o perpetuar una enfermedad autoinmune, como la AR. El papel crucial de MICA y NKG2D en el desarrollo de la AR permite especular que, como se ha demostrado en ratones diabéticos NOD25,26, el bloqueo de este mecanismo por ejemplo mediante la utilización de anticuerpos podría tener efectos terapéuticos beneficiosos para el tratamiento de la enfermedad.