


El síndrome de Camurati-Engelmann o displasia diafisaria progresiva es una patología que se caracteriza por la aparición de la hiperostosis de las diáfisis de los huesos largos (tibia, fémur, peroné…) que aparece de forma gradual y puede incluso afectar a las metáfisis, aunque característicamente nunca afecta a las epífisis. Además puede llegar a afectar a otros huesos, como el cráneo.
Se trata de una patología genética rara de herencia autosómica dominante, de penetrancia incompleta y expresividad variable. Se han descrito 200 afectados, de ambos sexos y de todas las razas.
Se debe a una mutación del gen que codifica el factor de crecimiento β-1.
Característicamente, la enfermedad comienza en la infancia y se caracteriza por la aparición de una marcha claudicante, dolor óseo, sobre todo en los miembros inferiores, y atrofia muscular y del tejido adiposo. Cuando el cráneo está afectado, pueden aparecer síntomas neurológicos, siendo el más frecuente la sordera.
Su diagnóstico se basa en los hallazgos clínicos más las alteraciones radiográficas típicas (ensanchamiento gradual e irregular de la diáfisis de los huesos largos). También puede ayudar en su diagnóstico la gammagrafía ósea. El diagnóstico de certeza se realizaría mediante un estudio genético de la mutación.
Los glucocorticoides son el tratamiento de elección de esta enfermedad. En el futuro, el tratamiento genético puede ser la cura de esta enfermedad.
Camurati-Engelmann disease (CED), or progressive diaphyseal dysplasia, is characterized by hyperostosis of the diaphyses of the long bones (tibiae, femora, humeri…) that appears gradually and can affect the metaphyses; the epiphyses, however, are characteristically spared. In addition, other bones, such as the skull, may be affected.
CED is a rare autosomal dominant genetic disorder. Penetrance is reduced and expressivity is variable. Over 200 patients of both genders and all races have been described.
CED is caused by a mutation in the gene encoding for transforming growth factor ß-1.
Typically, CED symptoms begin in childhood and usually consist of a waddling gait, bone pain in the lower limbs, muscular weakness and reduced subcutaneous fat. If the skull is affected, neurological symptoms can appear, the most common being hearing loss.
Diagnosis is based on clinical findings and typical radiographic changes (gradual and irregular thickening of the diaphysis of the long bones). Scintigraphy is also useful in diagnosis. Definitive diagnosis is provided by mutation analysis.
The main treatment for this disease consists of glucocorticoids. In the future, gene therapy may provide a cure for CED.
El síndrome o enfermedad de Camurati-Engelmann o displasia diafisaria progresiva es una patología autosómica dominante del grupo de las hiperostosis craneotubulares u osteocondrodisplasias.
La enfermedad fue descrita inicialmente por Cockayne en 19201; en aquella ocasión cuando se plantearon la etiología de la enfermedad se sugirió que el cuadro podría ser debido a una osteítis sifilítica. Camurati en 1922 fue el primero que sugirió que podría tratarse de una enfermedad hereditaria2 caracterizando hasta cuatro generaciones sucesivas con la misma enfermedad. Ya en 1929, Engelmann describió la forma típica grave3.
En 1948, Neuhauser propuso el nombre de displasia diafisaria progresiva, haciendo referencia a la naturaleza progresiva de la hiperostosis y la afectación de las diáfisis4, aunque el epónimo de Camurati-Engelman es ampliamente aceptado.
Esta enfermedad se transmite de forma autosómica dominante y afecta a todas las razas, aunque su penetrancia clínica y radiológica es bastante diversa.
El rasgo característico de este trastorno es la hiperostosis, que se produce de manera gradual, tanto en la superficie perióstica como endóstica de las diáfisis de los huesos largos; su afectación es simétrica y suele iniciarse en las diáfisis femoral y tibial, y extenderse a húmero, cúbito, radio... A veces, cuando la enfermedad progresa las metáfisis pueden afectarse pero las epífisis siempre están respetadas; en casos graves también puede afectar al cráneo, el esqueleto axial y la osteosclerosis es generalizada.
EtiologíaLa displasia diafisaria progresiva es una patología genética rara, que se transmite con una herencia autosómica dominante, con penetrancia incompleta y expresividad variable5.
El gen responsable de la displasia diafisaria progresiva fue localizado recientemente a nivel de la región 19q13.1-13.36.
Se han descrito un total de 24 familias portadoras de la enfermedad con alteraciones del gen que codifica el factor de crecimiento beta-1 (TGF-β-1)7.
El TGF-β-1 representa una proteína multifuncional de naturaleza polipeptídica que actúa sobre el crecimiento, la diferenciación y la morfogénesis celular. Sus receptores de membrana se han identificado en todos los tipos de células estudiadas. En el tejido esquelético, sirve como regulador sistémico que une la formación del hueso y la reabsorción del mismo a través de la regulación de la función de los osteoblastos y de los osteoclastos. Además, este factor de crecimiento también tiene acción inhibitoria en la miogénesis y la adipogénesis, lo que justifica los síntomas no óseos de la enfermedad8.
El gen que codifica el TGF-β-1 tiene 7 exones. Más del 90% de los individuos con enfermedad de Camurati-Engelmann tienen mutaciones identificables en el gen del TGF-β-1. La mayoría son mutaciones sin sentido en el exón 4 que provocan sustituciones de aminoácidos simples en la proteína codificada; el segundo exón más afectado es el exón 1. Los 3 alelos mutantes más frecuentes son: p.Arg210Cys, p.Arg218His, y p.Cys225Arg9.
El TGF-β-1es sintetizado a partir de una gran molécula precursora; este TGF-β-1 preproteína está formado por una proteína que contiene 278 aminoácidos que es proteolíticamente dividida en dos moléculas: el llamado péptido latente asociado (PLA) y el activo TGF-β-1. El PLA ayuda a la secreción y formación del TGF-β-1 en forma activa.
La mayoría de las mutaciones producen sustituciones de aminoácidos en el extremo carboxi-terminal del TGF-β-1 latente asociado al péptido. Estas mutaciones pueden alterar la dimerización de PLA y la unión de este al TGF-β1, lo que conlleva a un aumento de TGF-β1 activo en estos medios de comunicación celular en comparación con los fibroblastos normales10.
Se conoce que estas mutaciones y su naturaleza no se correlacionan con la severidad de las manifestaciones clínicas y radiológicas. Además, algunos individuos con mutaciones heterocigotas para el TGF-(1 presentan radiografías normales, por lo que la penetrancia exacta de la enfermedad no es bien conocida11.
Además, se piensa que puede existir un fenómeno de anticipación en su transmisión, al observarse la aparición de síntomas más graves y a edad más temprana en generaciones posteriores de algunas familias portadoras de la enfermedad, aunque este mecanismo todavía es desconocido.
FrecuenciaLa prevalencia de la enfermedad de Camurati-Engelmann es desconocida; se estima en 1 por cada 1.000.000 de habitantes. Se han identificado al menos 200 individuos. Todas las etnias y ambos géneros pueden estar afectados.
Se ha publicado una revisión de 41 familias procedentes de Europa, Asia, África, América, Australia y Oceanía, lo que nos demuestra que la enfermedad está ampliamente distribuida por el mundo.
En nuestro hospital, Hospital Universitario Virgen Macarena (Sevilla), se ha diagnosticado a 3 pacientes, hermanos entre ellos, afectados por dicha enfermedad; aunque no ha sido posible el estudio de su familia y genético de ellos, presentan todas las manifestaciones clínicas y radiológicas típicas de la enfermedad12.
Anatomía patológicaLas referencias histopatológicas identificadas, a pesar de existir cierta controversia, están representadas por las alteraciones a nivel de la cortical del hueso, donde se observa un aumento progresivo de la actividad de los osteoblastos en el lugar de la lesión ósea con ausencia de osteoclastos13, los cuales reaparecen tras el tratamiento glucocorticoideo14. En los músculos de los pacientes afectados se observa un adelgazamiento de las paredes de los vasos sanguíneos y cambios miopáticos que incluyen atrofia de las fibras tipo II15.
Recientemente se han publicado 2 casos (madre e hijo) de síndrome de Camurati-Engelmann donde se detectan cambios histopatológicos de osteomalacia en la biopsia, que parece estar relacionada con la mutación añadida del gen que codifica el RANK ligando16.
Manifestaciones clínicas17El curso clínico de la enfermedad puede ser muy diverso y en algunos pacientes parece producirse la remisión de los síntomas durante la edad adulta.
La enfermedad se presenta de forma específica durante la infancia, entre los 4 y 10 años de edad, suele progresar en la adolescencia y queda estacionaria o avanza lentamente en la edad adulta. La media de edad en los 199 casos comunicados es de 14 años, con un intervalo desde el nacimiento hasta los 76 años.
La enfermedad suele iniciarse con la aparición de una marcha claudicante o «de pato», dolor en las piernas, atrofia muscular progresiva y disminución de la grasa subcutánea en las extremidades, características que pueden confundirse con las de una distrofia muscular18.
La disminución de la masa muscular y la rigidez en las extremidades son más frecuentes en la zona proximal de los miembros inferiores, dificultando la bipedestación al estar sentado. Por ello, la marcha claudicante, que como hemos referido anteriormente es uno de los síntomas iniciales, aparece en el 64% de los individuos. Las contracturas articulares ocurren en el 43% de los individuos. El hábito marfanoide también ha sido descrito en algunos de los sujetos afectados.
En el 90% de los afectados se describe la presencia de dolor óseo. El dolor es descrito como constante y más intenso en los miembros inferiores. A menudo, el dolor aumenta con la actividad, el estrés o el frío. Muchos pacientes describen episodios o «crisis»’ de intenso dolor e incapacidad, cuya duración es indeterminada desde pocas horas hasta varias semanas.
El 52% de los pacientes afectados refiere dolor óseo a la palpación. También puede detectarse de forma intermitente inflamación, eritema y calor en las extremidades.
La susceptibilidad a las fracturas puede estar reducida debido al aumento de la densidad mineral ósea, pero la consolidación de estas, cuando ocurre, suele estar retrasada19.
Debido a estas alteraciones, los individuos gravemente afectados muestran un hábito corporal característico consistente en: megacefalia con frente prominente, exoftalmia, extremidades delgadas con huesos gruesos y masa muscular escasa. Suelen presentar un aspecto grácil, hábito marfanoide, marcha anadeante con ampliación de la base de sustentación, disminución de la fuerza con reflejos osteotendinosos exaltados, piernas arqueadas, pies planos y valgos, lordosis lumbar y escoliosis.
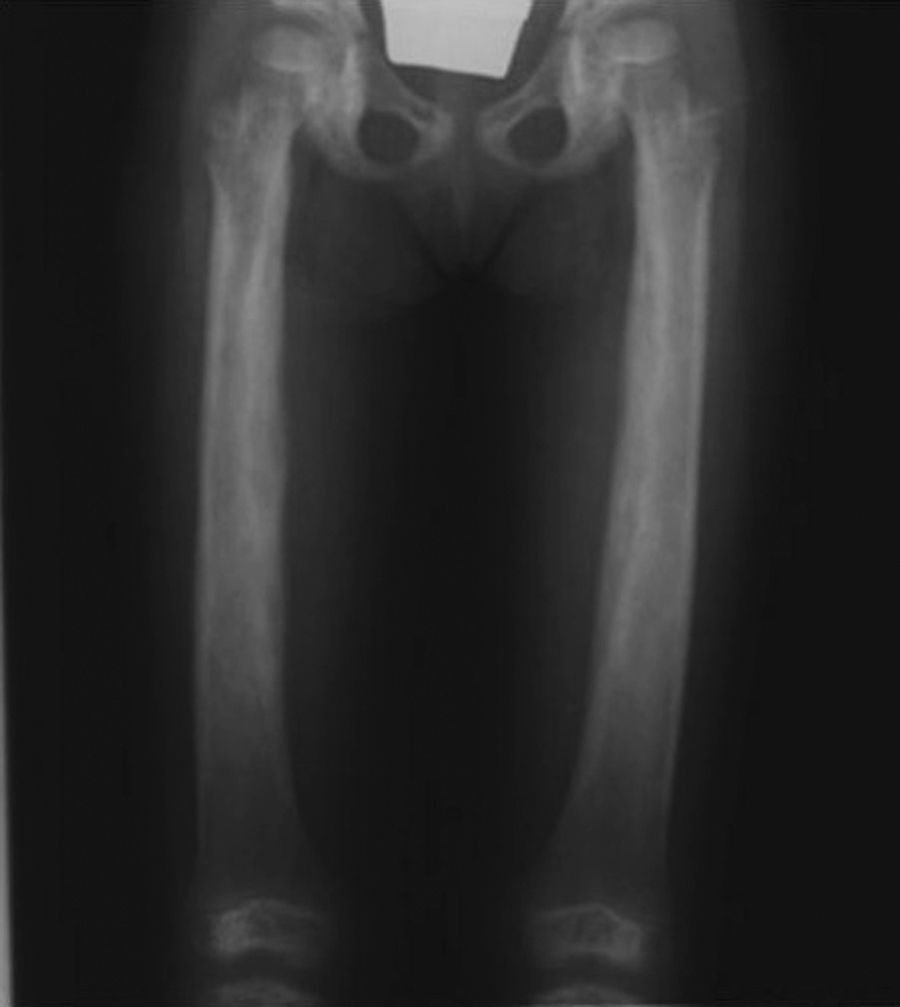
Alteraciones óseas20La característica más destacable del síndrome de Camurati-Engelmann es la hiperostosis de las diáfisis de los huesos largos, principalmente causada por la proliferación de hueso nuevo tanto en la superficie perióstica como endóstica, provocándose un ensanchamiento gradual de estos, que presentan superficies irregulares.
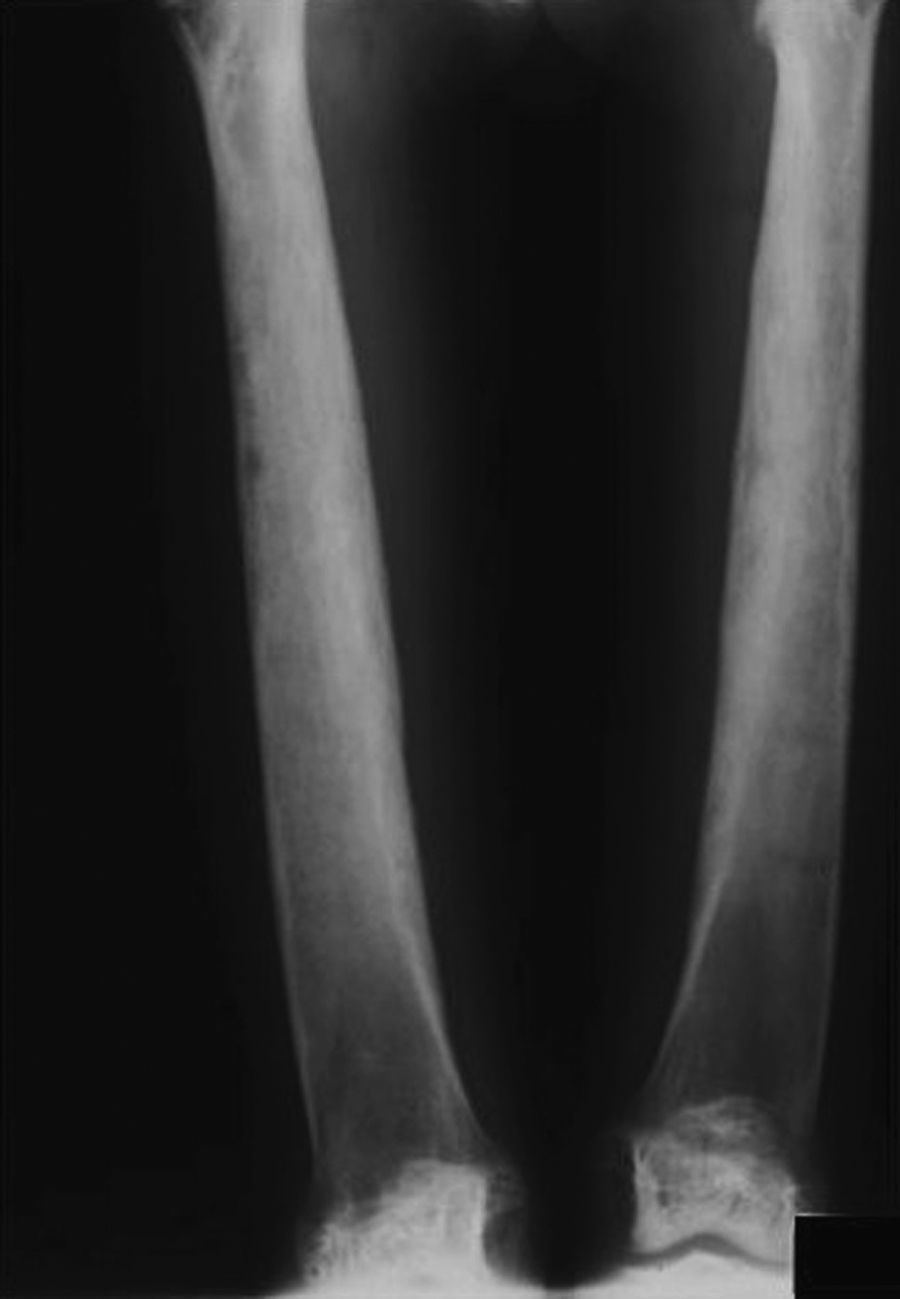
La esclerosis es simétrica y se extiende de forma gradual, pudiendo llegar a afectar a las metáfisis, pero nunca se afectan las epífisis. El canal medular muestra un estrechamiento en forma de frasco de Erlenmeyer.
Los huesos afectados por orden de frecuencia son: fémur, tibia, peroné, húmero, cúbito y radio. A veces, las tubulares de los huesos cortos, la base del cráneo, las mandíbulas, las escápulas, las clavículas y la pelvis también pueden afectarse. La columna vertebral está casi siempre íntegra. Ocasionalmente, se observan escoliosis grave y lordosis lumbar acentuada pero causada por la debilidad muscular.
Alteraciones neurológicasLa afectación craneal ocurre en un 38% de los casos. Cuando el cráneo se ve afectado pueden aparecer parálisis de los nervios craneales debido a la compresión o compromiso nervioso a distintos niveles como:
- –
Esclerosis del foramen óptico: puede producir edema de la papila y atrofia del nervio óptico, glaucoma y subluxación del globo ocular.
- –
Esclerosis del foramen del nervio facial provocando paresias y parálisis facial.
- –
Esclerosis del foramen interno acústico y del oído medio provocando hipoacusia de conducción o neurosensorial. Aproximadamente el 15% de los sujetos afectados presentan hipoacusia de conducción y/o neurosensorial. La pérdida conductiva puede ser causada por el estrechamiento del conducto auditivo externo, la afectación ósea de la cadena de huesecillos, el estrechamiento del conducto auditivo interno o la compresión del nervio coclear y/o su vascularización. La sordera neurosensorial puede ser causada por la compresión nerviosa.
- –
Esclerosis del foramen magnum, pudiendo producirse hiperreflexia, debilidad, paraparesia e incluso hasta muerte súbita21. Rara veces, clonus, pérdida de la sensibilidad, trastornos del habla, ataxia cerebelosa e incontinencia vesical e intestinal también han sido descritos.
- –
Alteraciones sistémicas.
Algunos pacientes, además de la sintomatología característica de la enfermedad, presentan síntomas orgánicos como hepatoesplenomegalia, fenómeno de Raynaud, anemia —que podría estar justificada por la disminución de la médula ósea roja—, leucopenia, atrofia cutánea, hiperhidrosis en manos y pies, retraso en la dentición, retraso en la pubertad, hipogonadismo..., lo que sugiere que el síndrome de Camurati-Engelmann no solo es una patología ósea y su manifestación es sistémica22.
DiagnósticoEl diagnóstico del síndrome de Camurati-Engelmann se basa en sus manifestaciones clínicas y en las alteraciones radiográficas.
En la evaluación inicial de esta enfermedad debe realizarse un examen neurológico, un estudio radiológico completo, medida de la presión arterial, un estudio auditivo, un estudio oftalmológico y un análisis general con reactantes de fase aguda.
Radiología: se caracteriza por un engrosamiento cortical de la diáfisis de aspecto fusiforme, que puede extenderse y afectar a las metáfisis pero nunca se afectan las epífisis. El canal medular se muestra estrecho (figs. 1-3).



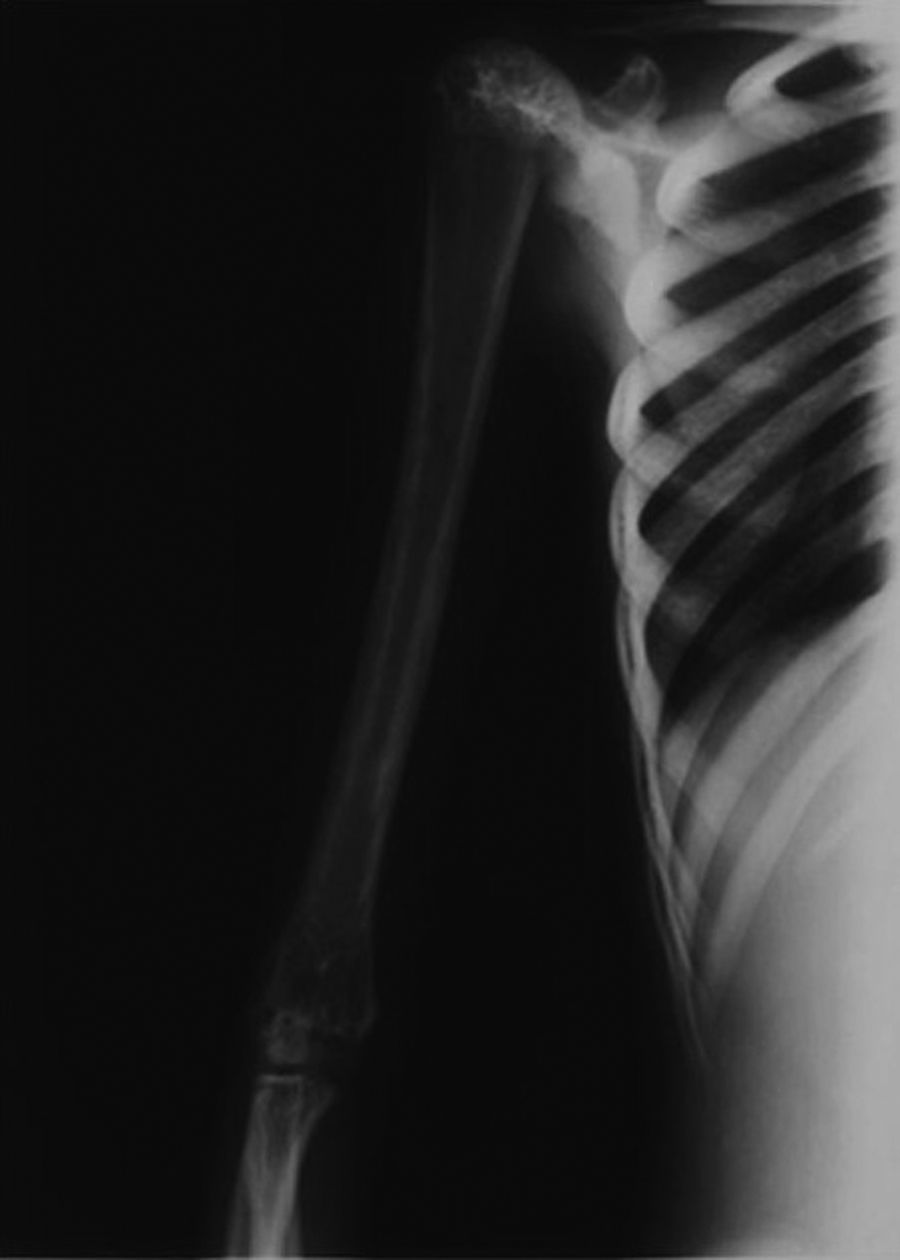
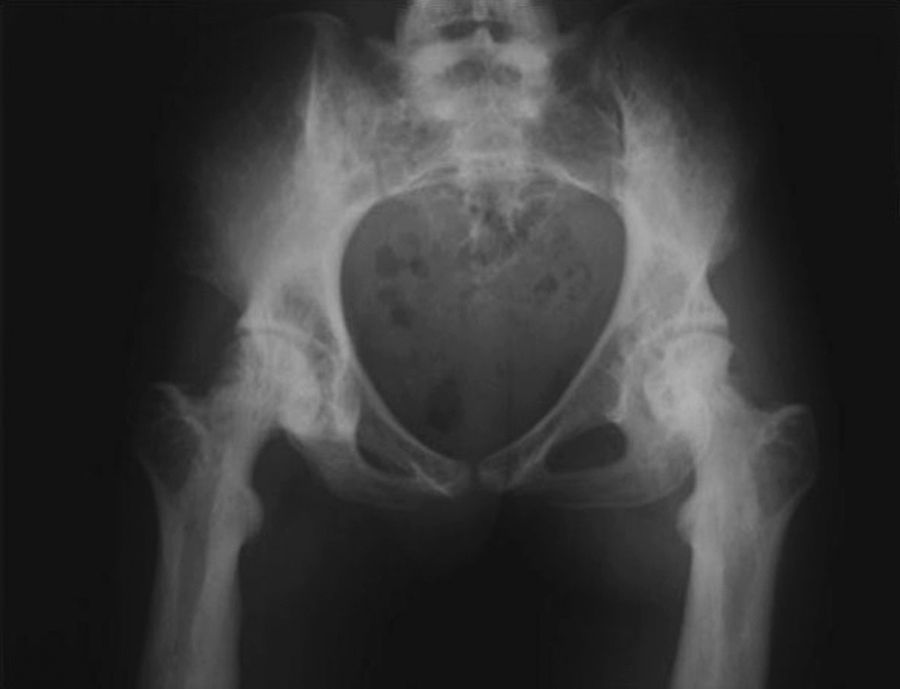
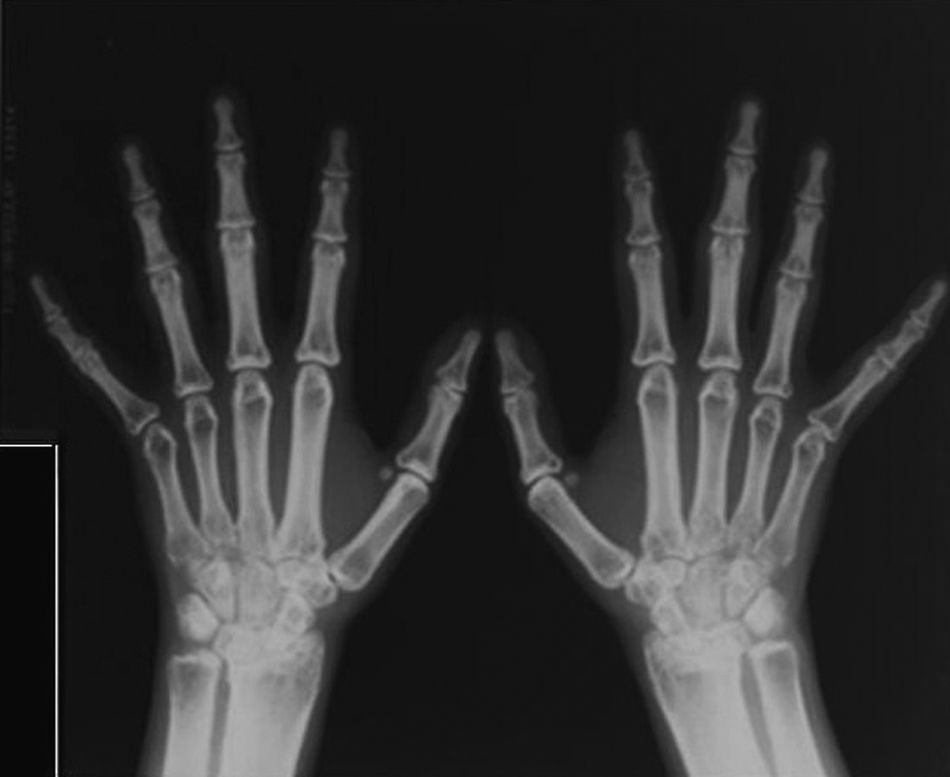
La imagen radiológica típica en los huesos largos aparece en el 94% de los pacientes con mutaciones identificables, un 54% también la presenta en el cráneo y un 63% en la pelvis (fig. 4)7. A veces, también existe afectación de huesos cortos, como el carpo o las falanges (fig. 5).


La imagen radiológica que se encuentra en la enfermedad de Camurati-Engelmann debe ser diferenciada de la displasia fibrosa, la fluorosis, la intoxicación por metales pesados, la osteodistrofia renal, la hipovitaminosis A, la sífilis, la osteomielitis y la osteopetrosis.
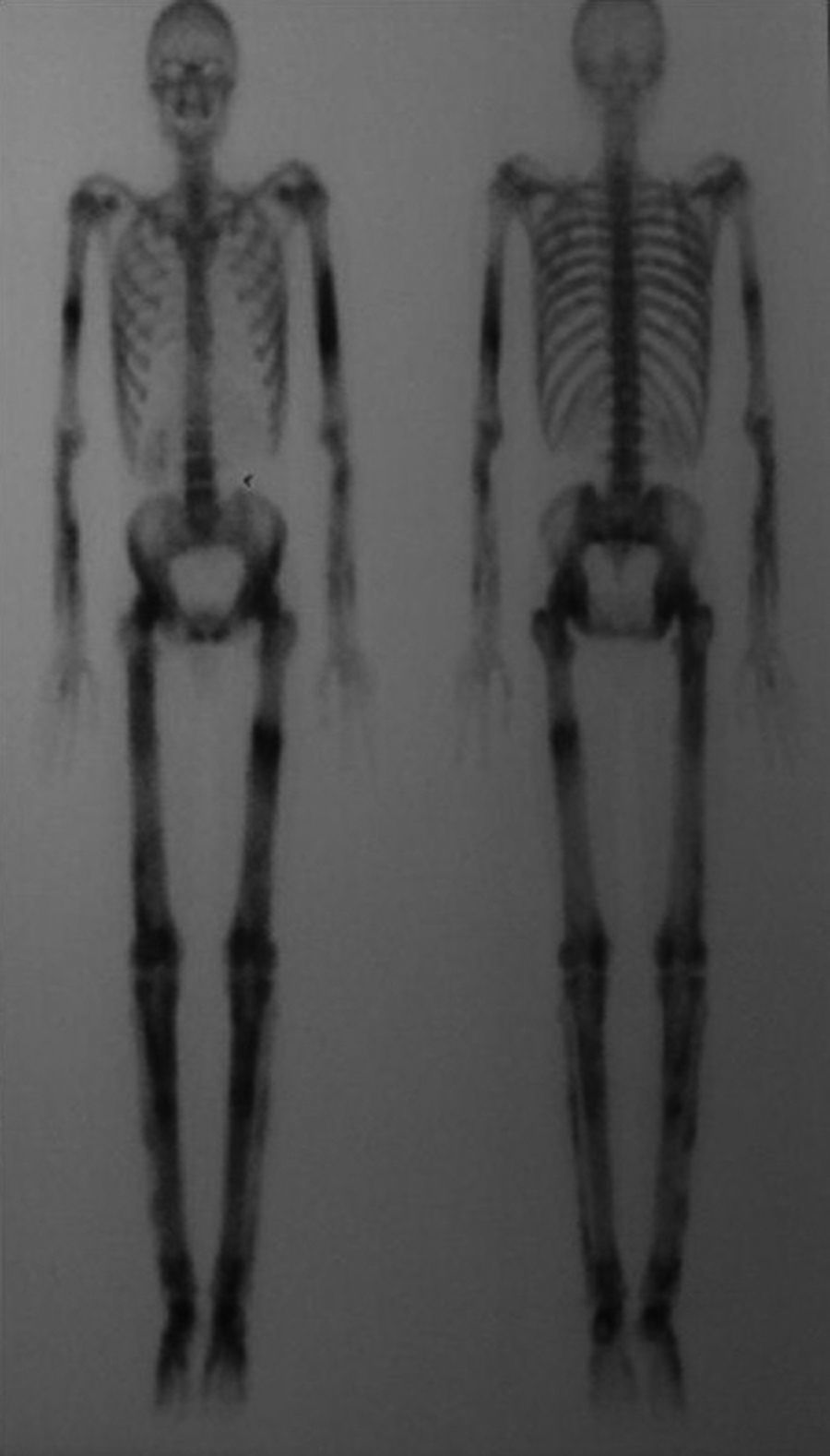
Gammagrafía ósea: detecta un aumento de la actividad osteoblástica en las regiones afectadas y esta actividad puede ser detectada incluso antes de la afectación radiológica (fig. 6)23.
 y de la pelvis.")
Laboratorio: no han sido identificadas referencias de laboratorio específicas de esta enfermedad; en algunos pacientes se ha reportado un aumento de velocidad de eritrosedimentación, un aumento de los niveles de fosfatasa alcalina, un incremento de excreción urinaria de hidroxiprolina y una reducción del nivel del hematocrito o de la hemoglobina.
Diagnóstico diferencialExisten varias enfermedades que provocan afectación esclerótica del tejido óseo, de las cuáles habrá que hacer el diagnóstico diferencial con el síndrome de Camurati-Engelmann. Entre ellas podemos destacar:
- –
Enfermedad de Ribbing: es una enfermedad osteoesclerótica de los huesos largos que es radiológicamente indistinguible de la enfermedad de Camurati-Engelmann; frecuentemente presenta dolor óseo después de la pubertad y se sabe que es causada por mutaciones en TGF-β-1, por lo que podemos concluir que ambas enfermedades son distintas variables fenotípicas de la misma enfermedad24.
- –
Displasia craneodiafisaria: que se caracteriza por un engrosamiento de los huesos de la línea media del cráneo provocando una ampliación del puente nasal e hipertelorismo ocular. La diferencia con el síndrome de Camurati-Engelmann es que la afectación del cráneo en esta es más leve, y si ocurre lo que provoca es prominencia frontal y proptosis. En cuanto a la afectación de los huesos largos, en la displasia craneodiafisaria esta se restringe a las diáfisis, mientras que en la enfermedad de Camurati-Engelmann las metáfisis pueden estar afectadas también.
- –
Síndrome de Kenny-Caffey tipo 2: se caracteriza por enanismo, engrosamiento cortical de los huesos largos, retraso en el cierre de las fontanelas, anomalías craneofaciales, hipocalcemia e hipoparatiroidismo. Ni anormalidades de laboratorio ni el retraso del cierre de las fontanelas ocurren en el síndrome de Camurati-Engelmann.
- –
Enfermedad de Paget juvenil: se caracteriza por la predisposición a fracturas, trabeculaciones y desviaciones en los huesos largos, características que no se presentan en el síndrome de Camurati-Engelmann.
- –
Displasia diafisaria con anemia: se caracteriza por una anemia severa y una susceptibilidad aumentada de infecciones. No hay evidencia de formación en el endostio ni en el subperiostio, lo que nos ayuda a distinguirla de la enfermedad de Camurati-Engelmann.
- –
Hiperostosis cortical generalizada: provoca un engrosamiento del endostio sin ampliación del eje diafisario. Hay también una afectación mandibular característica con aumento de la profundidad y del ángulo mandibular, que es distinta de la mandíbula ampliada; se encuentra solo en ocasiones en la enfermedad de Camurati-Engelmann.
- –
Displasia ósea esclerosante relacionada con SOST: incluye la esclerostosis y la enfermedad de Van Buchem. De la primera es característica la aparición de sindactilia, sobre todo del segundo y el tercer dedos; en la enfermedad de Van Buchem las manifestaciones son menores y la sindactilia no aparece. Estas enfermedades tienen una herencia autosómica recesiva, mientras que el síndrome de Camurati-Engelmann tiene una herencia autosómica dominante. En estas afecciones la hiperostosis presentan superficies lisas periósticas, mientras que los individuos con el síndrome de Camurati-Engelmann poseen un engrosamiento perióstico con una corteza áspera e irregular.
Lo más importante en los sujetos afectados por el síndrome de Camurati-Engelmann es la prevención de complicaciones secundarias.
Los glucocorticoides pueden retrasar la hiperostosis ósea y prevenir o retrasar la aparición de la participación del cráneo. Los estudios histológicos tras el tratamiento con glucocorticoides evidenciaron un aumento de la resorción ósea y de la remodelación secundaria debida al aumento de la actividad osteoblástica y la disminución del depósito de hueso laminar. Aunque existen autores que no refieren mejoría radiográfica y gammagráfica tras el tratamiento con glucocorticoides25, sí hay muchos otros que comunican una disminución de la esclerosis radiológica y una actividad disminuida por gammagrafía. En estudios de seguimiento a largo plazo, también se observó el éxito de esta terapia para prevenir la aparición de anemia, hepatoesplenomegalia, compresión pares craneales...
Se ha reportado un caso de mejoría del cuadro tras el embarazo, lo que nos habla de un posible efecto beneficioso para la enfermedad.
TratamientoGlucocorticoidesComo es ya conocido, el uso prolongado de glucocorticoides produce un efecto indeseable en el hueso, disminuyendo la masa ósea por varios mecanismos (apoptosis de osteoblastos, proliferación de osteoclastos, disminución de la absorción de calcio); este «efecto adverso» en pacientes con síndrome de Camurati-Engelmann se convierte en una ventaja, ya que ayuda a controlar el aumento de la formación ósea que se produce en esta enfermedad. Por otro lado, éstos parecen provocar un efecto directo positivo sobre la expresión y activación de TGF-β-1, cuyo mecanismo no es bien conocido, lo que podría ser un efecto adverso en los pacientes con sobreactivación del TGF-β-126. Además, se ha encontrado que los esteroides también interfieren en la regulación de la unión del TGF-β-1 al receptor de señal inhibiéndola, lo que podría ser beneficioso para el tratamiento de esta patología27.
Varios investigadores han comunicado la eficacia de los glucocorticoides en el tratamiento de la enfermedad. En todos los casos había una mejoría de los síntomas clínicos, como el dolor y la fatiga28. Además, como hemos referido anteriormente, producen una modificación de las alteraciones radiológicas.
En cuanto a las dosis utilizadas, en pacientes con síntomas graves pueden ser tratados con bolos de prednisolona de 1 a 2 mg/kg/día seguido de una rápida disminución de dosis hasta la mínima dosis eficaz. También pueden usarse dosis altas de corticoides en crisis álgicas. Los individuos menos sintomáticos pueden iniciar a 0,5-1 mg/kg/día. Es importante llegar a la dosis mínima eficaz para evitar, en medida de lo posible, los efectos secundarios de los corticoides, sobre todo en el crecimiento. Se describió que deflazacort, un derivado de la prednisolona, tenía un mismo efecto en la clínica y en los hallazgos radiológicos, pero con menos efectos adversos, por lo que podría ser un alternativa más segura29.
Algunos individuos presentan períodos quiescentes, donde puede discontinuar el tratamiento glucocorticoideo.
El tratamiento con glucocorticoides en pacientes asintomáticos no ha sido comunicado, pero debido a la variabilidad de la sintomatología y la disminución de la penetrancia, este tratamiento en individuos asintomáticos no parece estar recomendado.
LosartánAunque todavía no hay datos concluyentes, losartán puede ser utilizado en pacientes sintomáticos que no toleran corticoides o que padecen de hipertensión arterial. Este medicamento tiene un efecto anti-TGF-β-1 y está siendo probado en pacientes con síndrome de Marfan30.
BifosfonatosEl valor de los bifosfonatos en el tratamiento de esta enfermedad es discutido; existen pocos registros sobre el tratamiento con estos medicamentos y hay, por ejemplo, casos descritos en tratamiento con pamidronato o alendronato en que aumentan el dolor óseo y la actividad gammagráfica31 y otro con el mismo fármaco con mejoría32,33.
De todas formas, teniendo en cuenta que los bifosfonatos son fármacos antirresortivos ampliamente utilizados para incrementar la masa ósea en pacientes con osteoporosis, no parece que puedan tener un efecto prometedor para el tratamiento de esta enfermedad.
CalcitoninaEsta molécula es conocida por su potente efecto en la inhibición de la resorción ósea, por lo que conceptualmente no sería un fármaco útil en el tratamiento de esta enfermedad, aunque se han descrito efectos in vivo como inhibidor de la formación ósea34. Solo se ha descrito un caso de síndrome de Camurati-Engelmann tratada con calcitonina, donde se suspendió por no existir mejoría.
Otros fármacosAntiinflamatorios, analgésicos y otros métodos no farmacológicos son utilizados para el alivio del dolor, pero no han demostrado ningún cambio en la evolución de la enfermedad.
Tratamiento quirúrgicoEn cuanto al tratamiento de la hiperostosis de huesos largos, se han descrito algunos casos de limpieza del canal medular u osteotomías35, aunque estas lesiones pueden recurrir por la progresión de la enfermedad.
Por otra parte, en los síntomas centrales debidos a la hiperostosis del cráneo, como la sordera, puede realizarse una descompresión quirúrgica del conducto auditivo interno, pudiendo así mejorar la audición. Sin embargo, como la hiperostosis del cráneo es progresiva, la compresión del nervio craneal se repite a menudo. Existen pocos casos de enfermedad de Camurati-Engelmann con sordera y tratamiento efectivo de esta. Otras opciones quirúrgicas que se han descrito en estos pacientes son la miringotomía bilateral y el implante coclear, pero estos tratamientos son controvertidos, ya que la disminución del canal interno y la falta de funcionalidad del nervio son contraindicaciones para la colocación de un implante coclear36.
Terapia genéticaEn el futuro, la terapia genética podrá ser considerada como un camino hacia la curación de esta enfermedad. Se han realizado estudios in vitro en animales portadores de una mutación del gen del factor TGF-β-1, que presentaban altos niveles de este y hallazgos en la tibia típicos de esta patología, que fueron tratados con receptor inhibidor TGF-β-1 tipo I, modulando la actividad ósea y produciendo una mejoría de los hallazgos óseos37.
En cuanto al estudio génetico, estaría indicado siempre que se diagnostique a un individuo con esta enfermedad; es importante realizar un estudio a sus progenitores, radiológico y genético, ya que la mayoría de los afectados tienen a uno de los progenitores afectados, aunque a veces no es así, y la enfermedad es debida a una nueva mutación. En todos los casos es importante la realización del estudio genético, para poder aportar información a los sujetos afectados y sus familias sobre la historia de la enfermedad, el tratamiento y el modo de herencia, además de los riesgos de otros miembros de la familia de presentar la enfermedad.
Se puede realizar un diagnóstico prenatal mediante el análisis del ADN extraído de células fetales mediante una amniocentesis, y así poder conocer la presencia de la mutación causante de la enfermedad en el feto.
No obstante, como las manifestaciones clínicas y radiológicas no guardan relación con la mutación, e incluso existen pacientes con la mutación sin manifestaciones clínicas, el consejo genético se hace difícil, ya que con los datos actuales no podemos predecir con exactitud la afectación del paciente según la alteración genética presente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.