


El síndrome (o signo) del dedo azul (SDA) es la manifestación cutánea de un gran número de enfermedades que producen un compromiso isquémico agudo o subagudo en uno o más dedos. La causa más frecuente del SDA es la disminución del flujo arterial por el compromiso u oclusión de pequeños vasos periféricos, conservándose los pulsos distales palpables. La disminución del flujo arterial puede producirse por diferentes mecanismos patogénicos incluyendo la trombosis, la embolia, la vasoconstricción severa o la afección inflamatoria o no inflamatoria de la pared vascular. El dedo que sufre la isquemia adquiere un tono azul o violáceo, lo que da nombre al síndrome, y puede llegar a presentar fenómenos de necrosis.
Independientemente de la causa, este cuadro clínico es una urgencia médica que requiere un diagnóstico y tratamiento específico rápido, dado el riesgo de progresión a necrosis irreversible.
Blue digit syndrome (or sign) is a cutaneous manifestation of multiple diseases that produce acute or subacute ischemic compromise in one or more fingers or toes. The most frequent cause of this syndrome is a reduction in arterial blood flow due to compromise or occlusion of small peripheral vessels, with preservation of the distal pulses. The reduction in blood flow may be caused by a variety of pathogenic mechanisms including thrombosis, embolism, severe vasoconstriction, or inflammatory or non-inflammatory lesions of the vascular wall. The finger or toe affected by ischemia turns blue or violet and may develop necrosis.
Independently of the cause, blue digit syndrome is a medical emergency requiring rapid diagnosis and specific treatment, given the risk of progression to irreversible necrosis.
El compromiso isquémico agudo o subagudo de uno o más dedos es un escenario clínico al que ocasionalmente nos tenemos que enfrentar los reumatólogos ya que, en la mayoría de los casos, cursa con pulsos periféricos conservados por lo que inicialmente se suele descartar erróneamente una patología vascular. En la bibliografía de origen anglosajón se utiliza el término de síndrome del dedo azul (SDA) o de los dedos azules («Blue toe syndrome», «Blue finger syndrome» o «Blue digit syndrome») para designar a este cuadro clínico1–8.
El SDA puede afectar a un solo dedo, aunque con mayor frecuencia afecta a varios. Se pueden afectar tantos los dedos de la mano como los del pie y puede ser bilateral. La isquemia provoca dolor y un cambio de coloración en el dedo afecto que adquiere un tono azul o violáceo, lo que da nombre al síndrome. Una vez instaurada la isquemia se puede desarrollar ulceración, pérdida de tejido, infección y gangrena, siendo necesaria en ocasiones la amputación.
Aunque las primeras descripciones del SDA correspondían a casos producidos por la embolización de material aterotrombótico procedente de una placa arteriosclerótica (émbolos ateromatosos o émbolos de cristales de colesterol), los mecanismos patogénicos que dan lugar a este síndrome son variados e incluyen otras causas de disminución del flujo arterial, la disminución del retorno venoso y la hiperviscosidad sanguínea1–13. La importancia de conocer este síndrome o signo y sus diferentes etiologías radica en que un tratamiento rápido y específico evita la progresión a necrosis irreversible e incluso a veces puede salvar la vida del paciente.
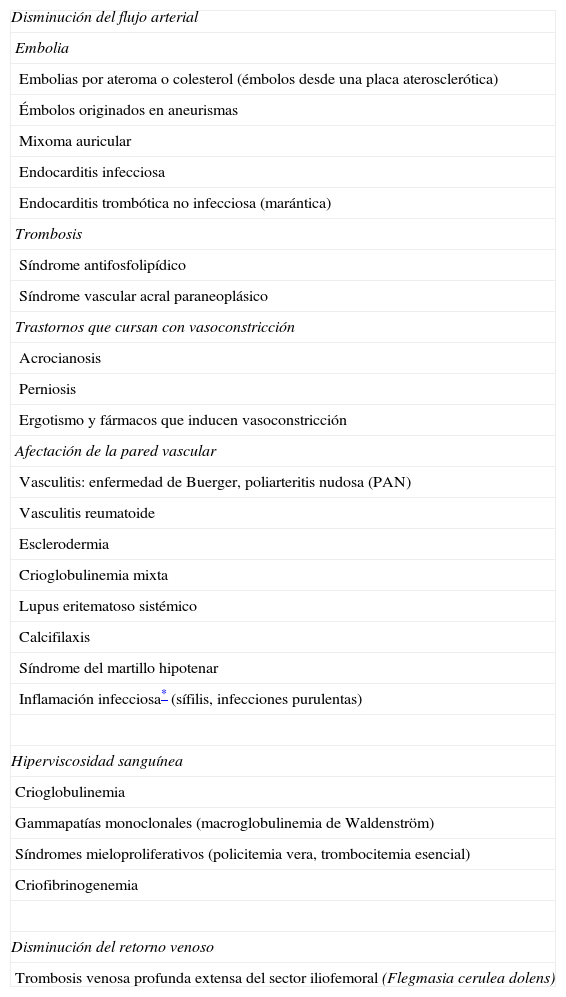
EtiologíaLas causas del SDA (excluyéndose el traumatismo previo o situaciones de cianosis generalizada) se recogen en la tabla 11–13. La más frecuente es la disminución del flujo arterial por el compromiso u oclusión de pequeños vasos periféricos; los mecanismos patogénicos que pueden producirlo incluyen la embolia, la trombosis, la vasoconstricción severa y la afección inflamatoria o no de la pared vascular.
Etiología del síndrome del dedo azul (SDA)
| Disminución del flujo arterial |
| Embolia |
| Embolias por ateroma o colesterol (émbolos desde una placa aterosclerótica) |
| Émbolos originados en aneurismas |
| Mixoma auricular |
| Endocarditis infecciosa |
| Endocarditis trombótica no infecciosa (marántica) |
| Trombosis |
| Síndrome antifosfolipídico |
| Síndrome vascular acral paraneoplásico |
| Trastornos que cursan con vasoconstricción |
| Acrocianosis |
| Perniosis |
| Ergotismo y fármacos que inducen vasoconstricción |
| Afectación de la pared vascular |
| Vasculitis: enfermedad de Buerger, poliarteritis nudosa (PAN) |
| Vasculitis reumatoide |
| Esclerodermia |
| Crioglobulinemia mixta |
| Lupus eritematoso sistémico |
| Calcifilaxis |
| Síndrome del martillo hipotenar |
| Inflamación infecciosa* (sífilis, infecciones purulentas) |
| Hiperviscosidad sanguínea |
| Crioglobulinemia |
| Gammapatías monoclonales (macroglobulinemia de Waldenström) |
| Síndromes mieloproliferativos (policitemia vera, trombocitemia esencial) |
| Criofibrinogenemia |
| Disminución del retorno venoso |
| Trombosis venosa profunda extensa del sector iliofemoral (Flegmasia cerulea dolens) |
De forma excepcional, en la sífilis se puede observar un SDA secundario a un infiltrado perivascular linfohistiocitario, junto con células plasmáticas y una hinchazón endotelial101. En pacientes que tienen una infección biogénica localizada en el pie o en la mano puede producirse un compromiso del flujo arterial que dé lugar a un SDA
El SDA también puede presentarse en el contexto de hiperviscosidad sanguínea, habiéndose descrito en pacientes con crioglobulinemia y, con mucha menor frecuencia, en las gammapatías monoclonales (fundamentalmente en la macroglobulinemia de Waldenström), síndromes mieloproliferativos (policitemia vera y trombocitemia esencial) y criofibrinogenemia14–22.
Muy raras veces el SDA es producido por una alteración del retorno venoso, observándose como manifestación infrecuente de una trombosis venosa profunda extensa del sector iliofemoral que compromete el retorno venoso y, secundariamente, el arterial por aumento de la resistencia al flujo de éste (esta situación se denomina también flegmasia cerulea dolens o inflamación azulada dolorosa)23–25.
Son muchas las enfermedades que pueden producir un SDA por disminución del flujo arterial (tabla 1). Dentro de ellas se incluyen algunas enfermedades autoinmunes sistémicas y las seudovasculitis. Como se comentará más adelante, las seudovasculitis son un grupo heterogéneo de enfermedades de etiología diversa que en ciertas ocasiones se las puede confundir con las vasculitis como, por ejemplo, cuando cursan con un SDA8–13. La frecuencia de estas enfermedades en las diferentes series de SDA varía según el especialista médico implicado. A modo de ejemplo, en un trabajo retrospectivo realizado en nuestro servicio que analizaba la frecuencia, etiología y evolución de los pacientes ingresados por SDA entre 1990 y 2008, las principales causas fueron las vasculitis (fundamentalmente vasculitis reumatoide, enfermedad de Buerger y poliarteritis nudosa), seguidas en frecuencia por la vasculopatía esclerodérmica y las seudovasculitis (responsables de un 14% de los casos)26.
Enfermedades autoinmunes sistémicas que causan con mayor frecuencia un síndrome del dedo azulTromboangitis obliterante o enfermedad de Buerger: es una vasculitis oclusiva que afecta a arterias de pequeño y mediano calibre de las zonas distales de las extremidades y, mucho más raramente, a vasos viscerales y cerebrales27–30. La enfermedad tiene una distribución universal, aunque es mucho más frecuente en Asia. Aunque se desconoce su etiología, se relaciona claramente con el consumo de tabaco (el 95% de los casos son fumadores). También parece existir una predisposición genética.
Afecta más a varones que a mujeres, en una relación de 9–1 en las series clásicas, aunque a medida que ha ido creciendo la proporción de fumadoras entre las mujeres, también ha crecido el porcentaje de afectadas por esta enfermedad. La mayoría de los pacientes son menores de 40–50 años. La anatomía patológica pone de manifiesto una vasculitis segmentaria que afecta a todas las capas del vaso y una trombosis inflamatoria oclusiva de los segmentos comprometidos.
Los síntomas iniciales más frecuentes (hasta en un 75% de los casos) son las lesiones de isquemia digital en las manos y, sobre todo, en los pies, que pueden evolucionar hasta la gangrena. En esta enfermedad los pulsos distales pueden estar ausentes y el test de Allen es positivo en la mayoría de los pacientes. También es frecuente el fenómeno de Raynaud, la claudicación intermitente de extremidades de predominio distal (siendo muy típica que afecte al pie o a los dedos de las manos), la tromboflebitis migratoria superficial y, en fases avanzadas, el dolor en reposo.
Las pruebas de laboratorio, incluyendo los reactantes de fase aguda y el estudio inmunológico, son estrictamente normales. El diagnóstico se suele confirmar mediante la arteriografía que, aunque no es patognomónica, resulta muy característica por la presencia de estenosis y oclusiones arteriales con abundantes colaterales alrededor de las mismas que corresponden a vasa vasorum muy dilatados (con un aspecto «en raíz de árbol» o «en sacacorchos» de los vasos afectados), en ausencia de lesiones ateromatosas y calcificaciones.
El tratamiento consiste en abandonar el consumo de tabaco (incluso la exposición pasiva o el uso de parches que contienen nicotina) y en medidas complementarias inespecíficas como antiagregantes plaquetarios, anticoagulantes, vasodilatadores, pentoxifilina y ejercicio físico. La utilización de inmunodepresores y glucocorticoides, agentes trombolíticos (estreptoquinasa o urokinasa intraarterial) o de estatinas es objeto de discusión; se han empleado poco y los resultados obtenidos son muy heterogéneos. El tratamiento con prostaglandinas por vía intravenosa (alprostadil o iloprost) es útil en pacientes con úlceras isquémicas. También se ha sugerido un posible efecto beneficioso de los antagonistas de los receptores de la endotelina-1 (bosentán). La simpatectomía se reserva para casos graves, progresivos y rebeldes al tratamiento médico. La revascularización quirúrgica se indica raramente, dado que la enfermedad afecta fundamentalmente a vasos pequeños. La amputación es el último escalón terapéutico27–30.
Poliarteritis nudosa (PAN): es una vasculitis necrosante sistémica que afecta a arterias de mediano y pequeño tamaño31–35. Las lesiones son característicamente segmentarias, con predilección por las zonas de bifurcación. Puede comprometer cualquier órgano (siendo el riñón y la piel los más involucrados en series de autopsia), excepto el pulmón que suele estar respetado. Predomina en los varones entre los 40–60 años, con una proporción de 1.5:1 en relación con las mujeres.
Las manifestaciones cutáneas están presentes en el 25–60% de los pacientes y consisten en livedo reticularis, púrpura palpable, nódulos subcutáneos, úlceras e isquemia digital que puede complicarse con gangrena. En ocasiones, la PAN puede quedar limitada a la piel como única expresión de la enfermedad36. Las manifestaciones generales (fiebre, afección del estado general y adelgazamiento) están presentes en la mayoría de los enfermos, y suele ser frecuente la afección renal (hasta en un 80% de los pacientes), la neuropatía periférica (75%) y la afección gastrointestinal (40%).
El diagnóstico se establece mediante confirmación histológica, generalmente a partir de biopsias superficiales (piel, músculo, nervio periférico o testículo). La biopsia de órganos internos (riñón, hígado) conlleva un riesgo elevado de hemorragia por rotura de un aneurisma o de desarrollo de una fístula arteriovenosa, por lo que únicamente deben realizarse cuando se consideren imprescindibles. De las diferentes lesiones cutáneas que provoca la enfermedad, los nódulos son las lesiones en las que con mayor probabilidad puede diagnosticarse la PAN. Deberían biopsiarse en su centro, con la profundidad suficiente para obtener una muestra de grasa subcutánea. Es importante tener en cuenta que las biopsias cutáneas en sacabocados no suelen ser útiles para confirmar el diagnóstico, puesto que pocas veces profundizan tanto como para alcanzar a los vasos culpables situados en la dermis profunda o en la grasa subcutánea y confirmar, de este modo, el diagnóstico de PAN. La única excepción son aquellos casos en los que la lesión cutánea es una vasculitis de un vaso de pequeño calibre (por ejemplo, una púrpura palpable o no palpable), en los que biopsia en sacabocados suele ser suficiente para los fines diagnósticos. Si se biopsia una úlcera, el mejor sitio no es el centro sino el borde. Las lesiones de la isquemia digital rara vez se biopsian, debido a que sus resultados son pobres y a los posibles problemas de cicatrización de las heridas32. Dado el carácter segmentario de la lesión, una biopsia negativa no excluye el diagnóstico y en ocasiones es necesaria su repetición. En estos casos, la angiografía selectiva del tronco celíaco y de las arterias renales puede ser de gran ayuda diagnóstica, al poner de manifiesto estenosis, irregularidades de la luz, trombosis y, en particular, microaneurismas, presentes hasta en un 70% de los pacientes (aunque no son patognomónicos de la PAN). Recordar que la PAN clásica es una enfermedad en la que los anticuerpos anticitoplasma de neutrófilo (ANCA) son negativos.
El tratamiento de la PAN clásica idiopática se basa en la administración de glucocorticoides, añadiéndose en los casos con enfermedad moderada-grave (con afección renal, gastrointestinal, cardíaca o neurológica), ciclofosfamida. En la mayoría de los pacientes suelen ser suficientes dosis de prednisona de 1mg/kg/día (máximo 60–80mg/día); en los casos con compromiso vital o mononeuritis múltiple grave se aconseja iniciar el tratamiento con pulsos de metilprednisolona de 1g diario intravenoso durante 3 días y seguir posteriormente con prednisona oral. La eficacia de la plasmaféresis en las formas graves de la PAN asociada al tratamiento inmunodepresor, es cuestionable. A diferencia de otros tipos de vasculitis, la PAN clásica es, a menudo, una enfermedad curable33–35.
Vasculitis reumatoide: la vasculitis reumatoide es indistinguible de la vasculitis necrosante de la PAN. Generalmente ocurre en pacientes varones con enfermedad erosiva de larga evolución, nódulos reumatoides, títulos altos de factor reumatoide e hipocomplementemia y denota peor pronóstico37,38. Las manifestaciones clínicas varían en función de los vasos afectados. La afección cutánea está presente en el 90% de los casos y su gravedad varía desde pequeños infartos sub- o periungueales o del pulpejo, hasta úlceras cutáneas profundas e isquemia digital que puede progresar a la necrosis. También es frecuente la neuropatía periférica (40% de los pacientes) en forma de mononeuritis múltiple o polineuropatía sensitivomotora, y las manifestaciones oculares, siendo las más importantes la escleromalacia perforante y la queratitis ulcerativa periférica. La afección renal y gastrointestinal (arterias mesentéricas) son, comparativamente, mucho menos frecuentes. También se ha descrito la afección coronaria (aunque la causa más frecuente de infarto de miocardio en los enfermos con artritis reumatoide sigue siendo la arteriosclerosis) y la afección pulmonar (en forma de hemorragia alveolar difusa o hipertension pulmonar)37,38. Siempre que sea posible se ha intentar obtener confirmación histológica del diagnóstico.
El tratamiento es bastante similar al de la PAN y se basa en la administración de glucocorticoides y, en casos con enfermedad moderada o grave, ciclofosfamida37,38. Como alternativas a la ciclofosfamida, en casos de contraindicación o fracaso terapeútico, se han documentado buenos resultados con azatioprina, antagonistas del factor de necrosis tumoral alfa (infliximab y etanercept) y rituximab37–46.
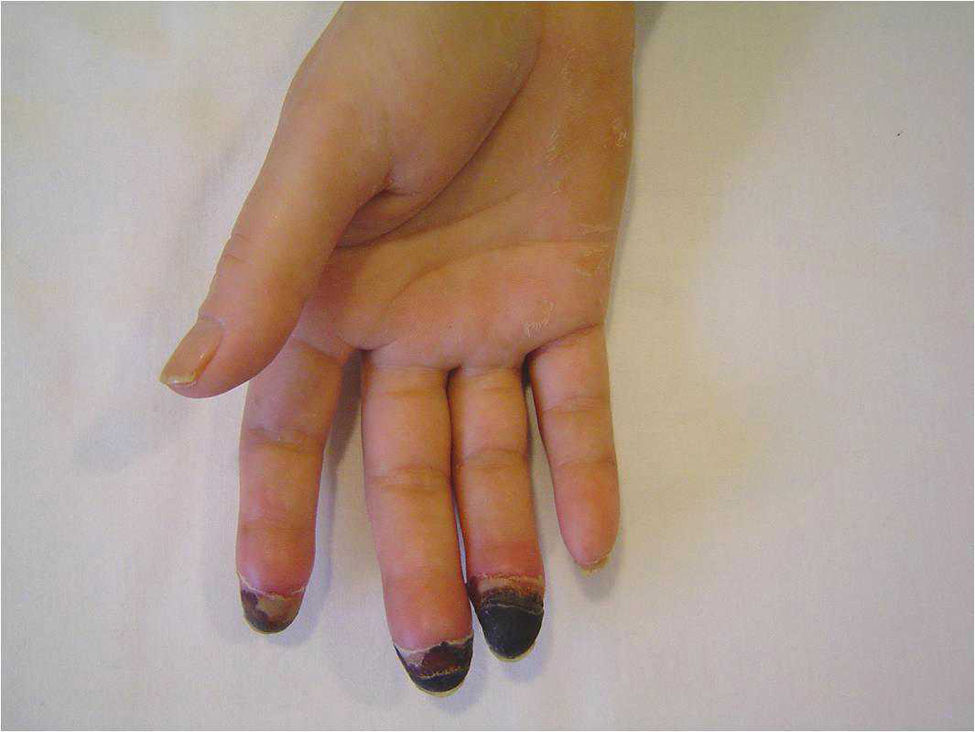
Esclerosis sistémica (esclerodermia): en la vasculopatía esclerodérmica se afecta la microcirculación (capilares) y las arteriolas, en las que se observa una necrosis capilar con hiperplasia de la íntima y fibrosis de la adventicia que acaban ocasionando la oclusión del vaso47. Clínicamente, las principales complicaciones vasculares periféricas de la esclerosis sistémica son el fenómeno de Raynaud, las úlceras isquémicas y finalmente, en casos de isquemia grave e irreversible, la necrosis digital y la consiguiente pérdida del dedo (fig. 1). La incidencia de amputación digital por paciente-año en los pacientes con úlceras isquémicas digitales se estima en un 1,2%48. En esta enfermedad es fundamental la identificación temprana de los síntomas y signos sugestivos de pre-isquemia. El dolor digital es el síntoma guía, siendo un dolor exquisito y de gran intensidad, que no suele calmar con la analgesia y que puede empeorar por la noche y con el roce. Los signos de pre-isquemia son una coloración azul-violácea del dedo que no se modifica con los cambios de temperatura y, a veces, la hinchazón. El tratamiento de la enfermedad vascular periférica se basa en la protección especial frente al frío y la administración de bloqueadores de los canales del calcio (como el nifedipino o el diltiazem). También han demostrado su utilidad los nitratos (tanto en parches de liberación sostenida como la nitroglicerina al 2% en gel de aplicación tópica), la doxazosina, el prazosin y el losartán (inhibidor específico del receptor tipo I de la angiotensina II), que pueden utilizarse combinados con los bloqueadores de los canales del calcio. En caso de úlceras isquémicas o necrosis se empleará el bosentán o la perfusión endovenosa de prostaglandinas (alprostadil o iloprost). Los antiagregantes plaquetarios (ácido acetilsalicílico o clopidogrel) pueden tener utilidad como terapia complementaria48.

Crioglobulinemia: es una enfermedad caracterizada por la presencia de crioglobulinas circulantes. Las crioglobulinas son inmunoglobulinas capaces de precipitar a temperaturas bajas. Las manifestaciones clásicas de la crioglobulinemia consisten en púrpura palpable, artralgias o artritis y astenia que ocurren en el 90–100% de los casos sintomáticos. Otras manifestaciones menos frecuentes son el fenómeno de Raynaud, las úlceras cutáneas, la isquemia digital, la neuropatía periférica y la glomerulonefritis membranoproliferativa difusa. La afección gastrointestinal y pulmonar es rara. Las manifestaciones clínicas de la crioglobulinemia aparecen como consecuencia de la oclusión vascular por crioprecipitados (más común en las crioglobulinemias de tipo I que cursan con criócrito elevado) y/o a consecuencia de una vasculitis por complejos inmunes formados por crioglobulinas (más frecuente en la crioglobulinemias mixtas)14,15.
En la crioglobulinemia mixta el tratamiento debe ser conservador en los casos leves. Cuando las lesiones cutáneas son importantes o existe afección visceral puede utilizarse prednisona a dosis de 0,5–1mg/kg de peso y día. En casos con lesión visceral grave puede asociarse plasmaféresis y/o inmunosupresores (ciclofosfamida, azatioprina o micofenolato). En la crioglobulinemia mixta asociada a infección por el virus de la hepatitis C la eliminación del virus es el objetivo del tratamiento. En pacientes con enfermedad leve-moderada se administrará interferon pegilado y ribavirina; en los casos con enfermedad grave se añadirá al tratamiento rituximab. En las formas fulminantes con compromiso vital se iniciará el tratamiento con plasmaféresis, glucocorticoides, rituximab y/o inmunosupresores, retrasándose el tratamiento con interferon pegilado y ribavirina a un segundo término, una vez superada la fase crítica del proceso49.
Lupus eritematoso sistémico (LES): la vasculitis es una complicación que se presenta en un 11–20% de los pacientes. Aunque la más frecuente es la urticaria vasculitis, también se han descrito casos de vasculitis necrosante sistémica tipo PAN y un tipo de vasculitis cutánea que afecta a pequeñas arterias y, que desde el punto de vista clínico, se limita a pequeños infartos en los pulpejos y el lecho ungueal de los dedos de manos y pies. Estas formas de vasculitis, en general, se asocian con lupus activo, hipocomplementemia, inmunocomplejos circulantes y títulos elevados de anti-ADN nativo50–53. Como se comentará más adelante, el síndrome antifosfolipídico que, con frecuencia se asocia al LES, también puede causar lesiones de isquemia digital54,55.
Seudovasculitis que pueden causar un síndrome del dedo azulLas seudovasculitis son un grupo heterogéneo de enfermedades de etiología diversa que en ciertas ocasiones se las puede confundir con las vasculitis8–13. La patogenia de estas entidades no es común; no obstante, el sustrato causante de las manifestaciones clínicas es el vaso sanguíneo. Son enfermedades relativamente frecuentes, cuyo diagnóstico es importante y que requieren un alto grado de sospecha. Conocer estas enfermedades evita un diagnóstico la mayoría de las veces tardío o, en el peor de los casos, erróneo, ya que muchas veces son confundidas con vasculitis y se instaura un tratamiento agresivo e innecesario (glucocorticoides e inmunodepresores) que comportan una importante morbilidad y que puede empeorar el curso de la enfermedad.
A continuación se revisan las principales seudovasculitis que se deben incluir en el diagnóstico diferencial del SDA.
Embolias por ateroma o colesterol: es una enfermedad producida por la oclusión de las pequeñas arterias por cristales de colesterol originados en la mayoría de los casos en las placas ateromatosas ulceradas de la aorta o en las arterias de ambas extremidades inferiores. Existen unos factores precipitantes, tales como los procedimientos percutáneos vasculares (es la causa más frecuente), la cirugía vascular, y el tratamiento con anticoagulantes o fibrinolíticos que debilitan la pared del trombo que cubre la placa de ateroma ulcerada56–63.
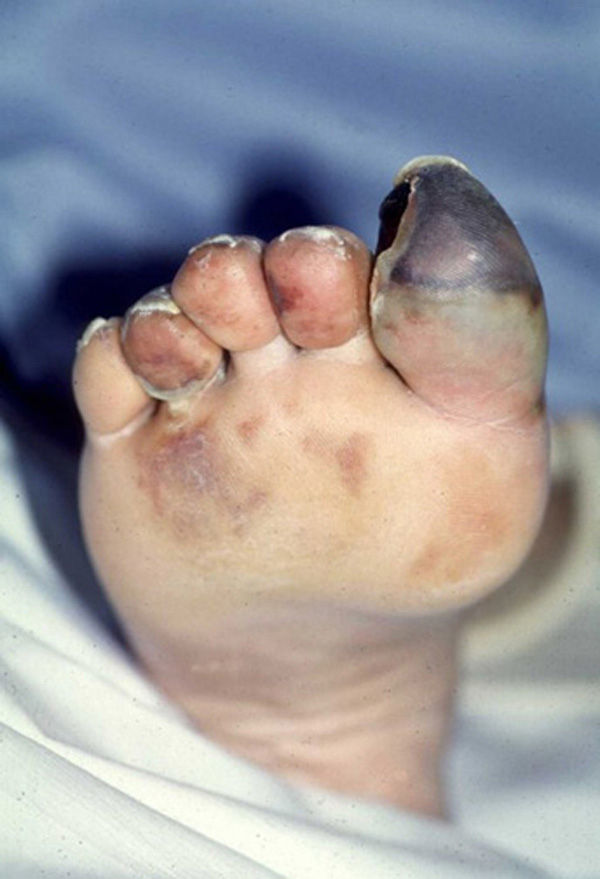
Las manifestaciones cutáneas habitualmente son las primeras y, con frecuencia, las únicas. Las más frecuentes son la isquemia digital que puede evolucionar hacia la ulceración y gangrena, la livedo reticularis y la púrpura (fig. 2). Suelen aparecer el día de la realización del evento causante del proceso o, con mayor frecuencia, unos días después y, en ocasiones, transcurridas unas semanas. En la mayoría de las ocasiones solo se observan en los pies, aunque también pueden presentarse en las piernas, las nalgas, el escroto y el pene. Dependiendo del origen de los cristales de colesterol, también se han descrito en el tronco y extremidades superiores. En las uñas se pueden observar hemorragias en astilla. En algunos pacientes la afección es multisistémica con compromiso renal (en forma de insuficiencia renal aguda, subaguda o incluso crónica, e hipertensión arterial acelerada y de difícil control), gastrointestinal (dolor abdominal o hemorragia, siendo infrecuente el infarto intestinal), ocular (por afectación de la circulación retiniana) y neurológica (amaurosis fugax, accidente vascular cerebral, coma o estado confusional y crisis epilépticas). En las pruebas de laboratorio destacan leucocitosis, eosinofilia (presente hasta en un 60% de los casos), aumento de los reactantes de fase aguda e hipocomplementemia. Los ANCA son negativos56–62.

La sospecha diagnóstica se realiza por los hallazgos clínicos y la anamnesis (siendo el dato fundamental el antecedente de intervención o exploración vascular), pero la confirmación definitiva se obtiene con el examen histológico de un órgano afectado. Las biopsias cutáneas de una gran variedad de lesiones, incluidas la livedo reticularis, la púrpura y áreas cianóticas, son diagnósticas en aproximadamente un 90% de los casos. En las biopsias fijadas con parafina, los cristales de colesterol se disuelven por las técnicas de preparación histológica dejando fisuras en forma de agujas en las pequeñas arteriolas con un diámetro de 100–200μm. Son cristales en aguja, biconvexos, situados en varios sentidos56–62.
El pronóstico es bueno en los casos en los que la enfermedad solo cursa con isquemia periférica, aunque un porcentaje de los pacientes acaba precisando la amputación de uno o varios dedos (tasa de amputación de hasta un 35–40% según las series). En cambio, en las formas con afección multisistémica el pronóstico es malo, con una mortalidad que alcanza el 70%. El tratamiento es sintomático y se basa en detener la progresión de la enfermedad con medidas agresivas de soporte, incluyendo el tratamiento con prostaglandinas intravenosas, la anticoagulación y/o antiagregación (a pesar de que se ha descrito su asociación etiológica), y la eliminación por vía quirúrgica o intraluminal de la causa de la embolización siempre que sea posible para conseguir la eliminación del foco embolígeno. La revascularización quirúrgica de los órganos afecto no suele ser posible, dado que la enfermedad afecta fundamentalmente a vasos pequeños63–65.
Además de la embolia desde una placa aterosclerótica, los trombos originados en los aneurismas también pueden embolizar a distancia y la clínica dependerá de la localización de dicho aneurisma66. La aterosclerosis es la causa más frecuente de los aneurismas.
Endocarditis: en las endocarditis infecciosas, además de la fiebre, es frecuente la presencia de manifestaciones extracardiacas que se producen por fenómenos embólicos o, en su defecto, mediadas por inmunocomplejos67,68. En la piel pueden aparecer lesiones en los dedos de manos y pies como petequias, los nódulos de Osler (pápulas dolorosas en el pulpejo de los dedos) y las manchas de Janeway (máculas no dolorosas en plamas y plantas). Por ello, la posibilidad de una endocarditis infecciosa debe tenerse en cuenta en pacientes con SDA y fiebre o soplo cardiaco (sobre todo, si además tiene esplenomegalia).
Otra causa potencial de SDA es la endocarditis trombótica no infecciosa (marántica)69–71. En estos casos, son trombos fibrinoplaquetarias en las válvulas cardíacas que suelen presentarse en el contexto de procesos malignos (los tumores más frecuentes son los adenocarcinomas de pulmón, páncreas o del tracto gastrointestinal), LES (endocarditis de Libman-Sachs) o síndrome antifosfolipídico. Son vegetaciones de pequeño tamaño y asientan con mayor frecuencia en las válvulas mitral y aórtica, por lo que pueden embolizar a arterias pequeñas, generalmente del bazo, riñón y de las extremidades, cursando muchas veces de forma asintomática.
Mixoma auricular: es el tumor cardíaco más frecuente. En el 75% de los casos se origina en la aurícula izquierda. Suelen ser pedunculados y estar unidos a la pared interauricular. Son más frecuentes en mujeres y suelen presentarse entre la tercera y la sexta décadas de la vida. La tríada clásica consiste en problemas cardíacos obstructivos (disnea, edema agudo de pulmón, insuficiencia cardíaca derecha y síncope), síndrome constitucional (incluyendo fiebre y artromialgias) y embolias. Los fenómenos embólicos ocurren en un 30–40% de los pacientes y suelen afectar al sistema nervioso central, circulación retiniana o producir isquemia periférica. El diagnóstico se establece mediante ecocardiografía. El tratamiento es quirúrgico. Presenta una tasa de mortalidad menor del 3% y las recurrencias alcanzan el 5%, sobre todo en los pacientes afectados por el complejo de Carney (mixomas, endocrinopatías y alteraciones cutáneas)72,73.
Síndrome antifosfolipídico: este síndrome imita las vasculitis porque puede presentarse de forma multisistémica o con una serie de manifestaciones cutáneas muy indicativas de vasculitis55,74–77. Las manifestaciones clínicas más frecuentes son la trombosis, tanto arteriales como venosas, y los abortos de repetición y/o muertes intrauterinas. Los fenómenos trombóticos más frecuentes son las trombosis venosas profundas en extremidades, que a menudo se complican con tromboembolias pulmonares, seguidas en frecuencia por las trombosis arteriales cerebrales. Las manifestaciones cutáneas más frecuentes son la livedo reticularis, la aparición de úlceras cutáneas localizadas sobre todo en las piernas, hemorragias en astilla y lesiones de isquemia digital que pueden evolucionar a la gangrena55. La tríada de livedo reticularis, accidentes vasculares cerebrales y presencia de de anticuerpos antifosfolípido se conoce como síndrome de Sneddon78. Algunos pacientes pueden desarrollar un cuadro trombótico multisistémico (fundamentalmente con afección renal, pulmonar, cardíaca y neurológica) de curso muy grave, denominado síndrome antifosfolipídico catastrófico79.
El diagnóstico se basa en conjugar las manifestaciones trombóticas con la presencia de anticuerpos antifosfolípido. El tratamiento en los pacientes que han sufrido una trombosis es la administración de anticoagulantes75–77. En la forma catastrófica además de la anticoagulación deben administrarse glucocorticoides y asociarse plasmaféresis79.
Síndrome vascular acral paraneoplásico: es el término acuñado para describir la asociación clínica de neoplasia, fenómeno de Raynaud y acrocianosis o gangrena de los dedos de las manos y los pies80. El 60% de las neoplasias son carcinomas, fundamentalmente adenocarcinomas de pulmón, ovario y estómago, casi siempre en estadios avanzados y con presencia de metástasis (41% de los casos). Un 20% son de origen hematológico y un 20% son sarcomas o cánceres de origen desconocido. Las lesiones cutáneas pueden preceder al diagnóstico del cáncer (45% de los casos). El mecanismo patogénico de este síndrome es aún poco conocido, habiéndose implicado la producción de sustancias vasoconstrictoras por las células tumorales y la embolia de pequeños trombos tumorales. El estudio histológico muestra necrosis fibrinoide y necrosis arterial y venosa.
Al margen del tratamiento sintomático (antagonistas del calcio, antiagregantes y prostaglandinas endovenosas), el cuadro mejora con el tratamiento de la neoplasia de base, habiéndose descrito la curación completa del síndrome en el 48% de los pacientes en los que se pudo curar el cáncer.
Acrocianosis: se trata de un síndrome vascular permanente, que afecta de modo preferente a las mujeres. Habitualmente se inicia en la pubertad, por lo general antes de los 20 años, y se caracteriza por la presencia de frialdad y una coloración azulada o eritrocianótica persistente en las partes distales de las extremidades en ausencia de dolor. La afección es bilateral y simétrica. Predomina en las extremidades superiores, en dedos y manos, y en menor grado en los dedos de los pies. La elevación de la extremidad disminuye la intensidad de la coloración azulada, que comienza a hacerse rojiza. El frío no actúa como desencadenante, sino como factor agravante de la sintomatología, que es permanente, si bien menos acentuada en épocas de calor. Existe siempre hiperhidrosis en la zonas palmares, en ocasiones muy intensa y agravada por situaciones emocionales.
La causa de este cuadro clínico parece ser la vasoconstricción de arterias y arteriolas de pequeño calibre, que origina una disminución del flujo, con ingurgitación de los capilares y vénulas subpapilares. El diagnóstico es fundamentalmente clínico.
El trastorno suele persistir durante toda la vida, aunque a veces mejora con los embarazos. El pronóstico es benigno, dado que no se complica con trastornos tróficos, úlceras o gangrena, aunque puede condicionar un problema estético, máxime cuando existe intensa hiperhidrosis. El tratamiento implica evitar los ambientes fríos y la administración de sustancias vasoactivas como pentoxifilina o buflomedil. También se han utilizado los inhibidores del calcio, los antagonistas de la serotonina y las prostaglaninas. La práctica de una simpatectomía quirúrgica no ha demostrado su utilidad81,82.
Perniosis: la perniosis (sabañones) es una enfermedad vascular producida por el frío. Se manifiesta con lesiones cutáneas en forma de pápulas, parches o placas eritematovioláceas, dolorosas y pruriginosas. Suelen aparecer horas después de la exposición al frío. Las lesiones se localizan en zonas acras: dedos de manos y pies, cara, nariz y pabellones auriculares83. Suelen ser bilaterales. En ocasiones tienen un curso crónico, con aparición de ampollas y úlceras. Los pulsos son normales. Se asocia a otros fenómenos precipitados por el frío, tales como la acrocianosis y el fenómeno de Raynaud. El diagnóstico suele ser clínico, siendo fundamental tener en cuenta la exposición al frío. Las características histológicas son edema dérmico e infiltrados linfocíticos perivasculares y alrededor de las glándulas ecrinas, acompañados de queratinocitos necróticos y, en ocasiones, de vasculitis linfocítica. El estudio de la perniosis puede sacar a la luz alguna enfermedad sistémica oculta hasta el momento, como el LES (perniosis lúpica). También se ha descrito el lupus pernio familiar o síndrome de Aicardi-Goutières, encefalopatía hereditaria grave y progresiva de inicio precoz, con microcefalia evolutiva que, en un 40% de los casos, presenta lesiones de perniosis desde el año de vida, que pueden originar isquemia digital84.
El tratamiento de la perniosis consiste en evitar el frío y la humedad, y proteger las manos y pies. Se pueden utilizar vasodilatadores así como glucocorticoides tópicos.
Ergotismo: el ergotismo era una enfermedad frecuente en la Edad Media (siendo denominada también como el fuego de San Antonio) causada por la contaminación del centeno por un hongo, el Claviceps purpurae. Hoy día el ergotismo se produce por el uso de derivados de la ergotamina, fármacos que se utilizan en el tratamiento de la migraña. Son potentes vasoconstrictores que pueden causar isquemia periférica, tanto de extremidades superiores como inferiores. En este caso los pulsos periféricos están muy disminuidos o ausentes debido a la intensa vasoconstricción. También se han descrito isquemias abdominales. El diagnóstico se establece mediante la anamnesis y la reversibilidad de las lesiones al suspender el tratamiento. En casos graves son útiles las prostaglandinas intravenosas85,86.
Otros fármacos que pueden originar ergotismo o acrocianosis son el ritonavir, indinavir, anfotericina B deoxicolato, eritromicina, gemcitabina, imipramina, mesilato de pergolida y el interferon alfa-2A. Los fármacos vasopresores sistémicos (dopamina, noradrenalina y fenilefrina) pueden producir también acrocianosis, isquemia e incluso gangrena.
Calcifilaxis o arteriolopatía calcificante urémica: es una enfermedad infrecuente caracterizada por la ulceración isquémica de la piel como consecuencia de la calcificación de la capa media de las pequeñas arteriolas subcutáneas. Esta calcificación se asocia a una proliferación endotelial y fibrosis de la íntima, originando una oclusión luminal completa87–93.
Generalmente suele presentarse como complicación rara en pacientes con insuficiencia renal crónica en programa de diálisis, aunque también se han descrito casos en enfermos con insuficiencia renal sin programa de diálisis o trasplantados con disfunción del injerto. Asimismo, se han documentado casos aislados no asociados a insuficiencia renal en pacientes con artritis reumatoide, LES, sarcoidosis, hiperparatiroidismo primario, enfermedad de Crohn, hepatitis autoinmune y hepatopatía alcohólica89. El mecanismo etiopatogénico de la enfermedad es poco conocido. En los pacientes con nefropatía se han identificado como principales factores de riesgo para el desarrollo de esta complicación la hiperfosforemia mantenida, un producto calciofósforo elevado, el hiperparatiroidismo severo y la administración de quelantes cálcicos del fósforo y de derivados de la vitamina D para el control del hiperparatiroidismo secundario. En otros estudios también se han implicado como factores de riesgo el sexo femenino, la obesidad y el síndrome metabólico, un pobre estado nutricional, la diabetes mellitus, el hábito tabáquico, la enfermedad hepática, los corticoides sistémicos y el uso de anticoagulantes orales.
Según el área de presentación de las úlceras isquémicas, actualmente se tiende a distinguir 2 formas clínicas de calcifilaxis: la calcifilaxis acra, en la que las lesiones se localizan fundamentalmente en la zona distal de las extremidades superiores e inferiores, y la calcifilaxis proximal, con lesiones localizadas en la zona proximal de extremidades, tronco y nalgas, generalmente en áreas de gran adiposidad. La afectación distal puede comenzar en forma de livedo reticularis o cianosis acral, incluido el SDA, pero progresa de forma característica a la ulceración y la gangrena y, en muchos casos, a la amputación. Otras complicaciones son la necrosis peneana y la necrosis lingual.
El diagnóstico es fundamentalmente clínico; en los casos en los que se realiza biopsia se objetiva, al margen de los cambios vasculares anteriormente descritos, fenómenos inflamatorios en el panículo adiposo de distribución septal. El pronóstico es malo, con una mortalidad elevada sobre todo en los casos con lesiones proximales, fundamentalmente por la aparición complicaciones sépticas.
El tratamiento de la calcifilaxis no está bien establecido y aunque se han descrito múltiples intervenciones con potencial terapéutico, ninguna de ellas es uniformemente efectiva. Se han empleado con resultados muy heterogéneos el tiosulfato sódico intravenoso (por su acción antioxidante y quelante del calcio), bifosfonatos, sevelamer (quelante de fosfato), pentoxifilina, cinacalcet (modulador de los receptores de calcio situados en las glándulas paratiroides que reduce las concentraciones séricas de parathormona), vitamina K, tratamiento con oxígeno hiperbárico, uso de larvas estériles de Lucilia sericata para curar las úlceras, e incluso la paratiroidectomía urgente en los casos con hiperparatiroidismo grave. Siguen siendo fundamentales las medidas complementarias incluyendo la corrección de las alteraciones del metabolismo fosfocálcico, retirar los suplementos de calcio y vitamina D, aumentar la frecuencia de las diálisis a 6–8 sesiones semanales disminuyendo la concentración de calcio del baño de diálisis, el control de la infección, el desbridamiento quirúrgico o exéresis de las lesiones y la amputación de las extremidades en caso necesario87–94.
Síndrome del martillo hipotenar: es una enfermedad laboral que se produce como consecuencia de los traumatismos repetidos sobre la arteria cubital en el canal de Guyon. Se observa fundamentalmente en trabajadores manuales que golpean reiteradamente con la palma de la mano, aunque también se ha descrito en deportistas de voleibol, balommano, tenis o beisbol.
La fisiopatología se este síndrome viene determinada por la distribución anatómica de la arteria cubital en su entrada en la mano a través del canal de Guyon, el cual limita por su lado medial con el hueso pisiforme, lateral con el gancho del hueso ganchoso y dorsal con el ligamento transverso del carpo. En los 2 centímetros entre la rama profunda de la arteria cubital y el origen del arco palmar superficial, la arteria cubital solo está protegida por la piel, el tejido celular subcutáneo, el músculo palmar menor y la aponeurosis superficial. Al golpear sobre la eminencia hipotenar la arteria cubital choca contra el hueso ganchoso que hace de yunque, constituyendo un lecho duro para cualquier impacto sobre el vaso. Como consecuencia de los traumatismos repetidos se produce vasoespamo, lesiones en la íntima y disrupción de la media lo que favorecerá la agregación plaquetaria, la trombosis y la degeneración anurismática.
Del aneurisma se desprenden pequeños trombos que embolizan las arterias digitales, siendo el cuarto y quinto dedos de la mano dominante los más frecuentemente afectados. La oclusión de la arteria producirá en un momento dado una isquemia digital, aunque también puede manifestarse en forma de fenómeno de Raynaud. En esta ocasión los pulsos cubitales estarán abolidos y el test de Allen será positivo. Estudios recientes señalan que existe una displasia arterial predisponente en la mayoría de los pacientes. El diagnóstico se confirma mediante angiografía, angiotomografía axial computarizada con reconstrucción o angiorresonancia magnética. El tratamiento es conservador (analgesia, antiagregantes y prostaglandinas intravenosas) o quirúrgico en función del estadio clínico de las lesiones95–100.