


Ulomoides dermestoides (Fairmaire, 1893) is a beetle used in alternative medicine treatments in some South American countries. The objective of this study was to evaluate the cytotoxic and genotoxic effects of phenolic fractions (PF) from U. dermestoides. The PF were separated from crude extracts in acetone (PFAc) and ethanol (PFEtOH). The total phenolic content (TPC) was determined by Folin-Ciocalteu test. Volatile and semi-volatile compounds presents in crude extracts were identified by GC-MS; moreover, phenolic fractions were analyzed by HPLC-MS. The cellular viability, after exposition to phenolic fractions, was determined by Trypan blue exclusion test and MTT reduction assay on immortalized human keratinocyte cell line (HaCat); the degree of DNA damage was detected by alkaline comet-DNA assay. The TPC in PFAc and PFEtOH were: 11.34±0.88 mgGAE/g and 6.52±1.28 mgGAE/g, respectively (mean.dif: 4.951; p value = 0.0000). In both samples, HPLC-MS showed a pseudo-molecular ion [M-H]− at m/z 153, tentatively identified as protocatechuic acid. The results of cytotoxic assays suggest that the viability of HaCat cells depends on the concentration and exposure time of each treatment. Furthermore, the comet assay revealed moderate genotoxic effect after 48hours of exposure to PFAc (40 to 160μg.mL−1); cytotoxic/genotoxic activity of this fraction could be related to the higher phenol contents.
Ulomoides dermestoides (Fairmaire, 1893), es un escarabajo usado en medicina alternativa en algunos países de Sudamérica. El objetivo de este estudio fue evaluar el efecto citotóxico y genotóxico de fracciones fenólicas (FF) de extractos de U. dermestoides. Las FF se separaron desde extractos acetónicos (FFAc) y etanólicos (FFEtOH). El contenido de fenoles totales (CFT) se determinó mediante ensayo de Folin-Ciocalteu. Compuestos volátiles y semi-volátiles, presentes en los extractos crudos, se identificaron mediante CG-EM; por otra parte, las FF se analizaron por HPLC-EM. La viabilidad celular, después de exposición a las FF se determinó mediante la prueba de exclusión con azul de tripano y el ensayo de reducción con MTT, usando la línea celular de queratinocitos humanos inmortalizados (HaCat); el grado de daño en el ADN se detectó mediante el ensayo de ADN-cometa alcalino. CFT en FFAc y FFEtOH fueron: 11,34±0,88 mgAGE/g y 6,52±1,28 mgAGE/g, respectivamente (dif.media: 4,951; p value = 0.0000). En ambas muestras, HPLC-EM mostró un ion pseudo-molecular [M−H]− a 153m/z, identificado tentativamente como ácido protocateuico. Los resultados de los ensayos de citotoxicidad sugieren que la viabilidad de células HaCat depende de la concentración y el tiempo de exposición a cada tratamiento. Además, el ensayo cometa reveló efecto genotóxico moderado después de 48h de exposición a FFAc (40 a 160μg.mL−1). La actividad citotóxica/genotóxica de esta fracción podría estar relacionada con el contenido más alto de fenoles.
Around the world, many cultures use insects and their products as nutraceutical1. The Ulomoides dermestoides Fairmaire, 1893 (Coleoptera, Tenebrionidae, Diaperinae) (synonymy: Martianers dermestoides; Palembus dermestoides) is a darkling beetle of Asian origin, known in oriental culture for its aphrodisiac and therapeutic properties2; in South America, this beetle is eaten alive as an alternative therapy for bronchial asthma, psoriasis, vitiligo, chronic skin diseases, rheumatoid arthritis, hemorrhoids, diabetes mellitus, inflammation and different types of cancer3,4.
During the last decade, the interest to research the therapeutic effects attributed to the U. dermestoides has risen. Santos et al., 2010 showed the anti-inflammatory properties of polar extracts of this beetle, using the carrageenan-induced edema test in mice5; Crespo et al., 2011 described cytotoxic and genotoxic effects of dichloromethane extracts from whole body of U. dermestoides and the main quinone in their defense secretion (1,4 -benzoquinone), on cellular line of lung cancer (A549)6. Tobón et al., reported depressive activity on the central nervous system (CNS) of albino rats (Mus musculus Linnaeus, 1758) because of administration of oil extracted from U. dermestoides (3mg.kg-1, orally)7.
Additionally, presence of secondary metabolites with antioxidant activity8 and antioxidant enzymes such as superoxide dismutase9,10 on whole body U. dermestoides extracts has been described. This is proof of existence of complex systems anti-free radical in the beetles, that could be explored in the search of novel therapeutic agents for treatment of human disease associated with oxidative stress. However, it is also necessary to know the effect of U. dermestoides extracts or their fractions, on viability of healthy human cells. Skin is an important barrier in protecting the body from external chemicals and HaCat cell are an in vitro model to investigate cytotoxic effects on epidermal tissues11–13. In addition, HaCat cells exhibited a remarkably stable genetic balance over extended culture periods, without shifting to the tumorigenic phenotype14, for these reasons HaCat cell in vitro were selected for this research.
The objective of this study was to investigate cytotoxic and/or genotoxic effects of phenolic fractions from U. dermestoides whole body extracts, on a line of immortalized human keratinocytes (HaCat cells).
MATERIALS AND METHODSSample. A culture of U. dermestoides was established from a broodstock whose taxonomic identity was validated in the entomology department of the Natural Science Institute of the Universidad Nacional, Bogotá, Colombia (collection code ICN-45905). The beetles were bred under controlled temperature conditions (27 ± 2°C) and relative humidity (70-75%) and were fed exclusively with wheat bread and wheat bran. After 90 days of culture, adult individuals were separated from the substratum and were stored at -70°C.
Materials. Chemicals and solvents were obtained from Merck (Darmstadt, Germany) unless stated otherwise. Analytical grade chemicals were used. Solvents used in HPLC-MS, extraction and sample preparation were of HPLC grade.
Extracts. The frozen beetles were ground by friction with a mortar under a stream of liquid nitrogen until obtaining a pulverized material. Four grams of this material were mixed with 40mL of extraction solvent, the mixture was submitted to constant agitation at 4°C, during 48h. The extraction solvents used were: absolute ethanol (EtOH), butanol (BuOH), acetone (Ac) and ethylic acetate (EtAc). Homogenates were then clarified using a fritted funnel and centrifuged at 3,500g for 20min at 4°C in a Beckman Refrigerated Benchtop equipment (model GPR), the supernatants were stored in amber glass vials at -20°C.
Phenolic fractions (PF). Separation of the PF was done with column chromatography (40 x 2cm), using 20-60 mesh Amberlita® XAD®- 2 resin (Supelco, 10357), with a solvent flow rate of 10mL.min−1. It was added to a 1mL column of the total extract, and then two washes were done with 10mL of distilled water; compounds were eluted from the column with 10mL of methanol (MeOH)15. Eluates were lyophilized at - 40°C and 1.33 mBar, in a FreeZone 1-liter Benchtop Freeze Dry System (Labconco, Kansas City, MO).
Total phenolic content (TPC). The TPC was determined with Folin-Ciocalteu method described by Ozgen et al., 200816. Samples were prepared by dissolving the lyophilized phenolic fractions to a concentration of 0.2mg.mL−1. A volume of 2mL of this solution was mixed with 2.5mL of the Folin-Ciocalteu reagent (Panreac Qui¿mica S.L.U., Barcelona, Espan¿a), diluted at 10% v/v in distilled water. The mixture was incubated at room temperature for 2min, then 2mL of 7.5% w/v sodium carbonate were added, and the mixture was then incubated at 50°C for 15min. Absorbance of the sample was read in a Thermo Scientific Genesys 6 spectrophotometer (Madison, WI, USA) at a 765nm wavelength, the results were interpolated in a gallic acid (Sigma, G7384) standard calibration curve (0.1 – 0.00312mg.mL−1) and were expressed as milligram of Gallic Acid Equivalent (GAE) by gram of dry weight (mg GEA/g). The calculation was done using the following equation:
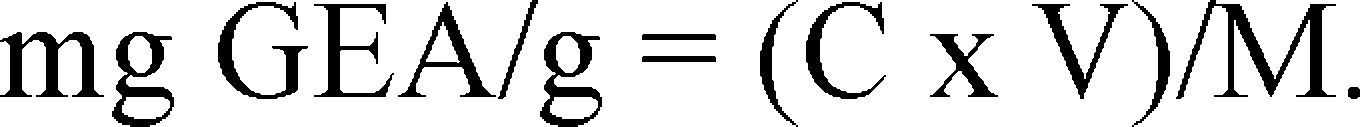
Where:
C = Concentration, determined by the gallic acid standard curve (mg.mL−1).
V = Volume of sample used in the test (mL).
M = Mass of the dry weight (g).
GC-MS. This procedure was performed at the Chromatography and Mass Spectrometry Laboratory of the Universidad Industrial de Santander. Preparation of the sample (total extracts of U. dermestoides) was carried out by direct injection of the samples into the chromatography equipment. The analysis was done in an AT 6890 Plus Series (Agilent Technologies, Palo Alto, California, USA.) gas chromatographer, coupled to a selective mass detector (Agilent Technologies, MSD-5975) operated in full scan mode. The columns that were employed in the analysis were DB-5MS (J & W Scientific, Folsom, CA, USA.) [5%phenyl-poly (dimethylsiloxane), 60 m x 0.25mm x 0.25μm]. The injection was carried out in Split mode (30:1), Voliny = 2μL. Tentative identification of registered compounds in the samples was established based on their mass spectrum (EI, 70Ev), using Adams databases, Wiley 138 and NIST 2005 (W*/N05)17.
HPLC-MS. The analysis was performed at the Mass Spectrometry Laboratory at the Universidad de Antioquia (University Research-SIU). Phenolic fractions from U. dermestoides extracts, were analyzed on an Agilent 1100 series LC-MS (Agilent Technologies, Waldbronn, Germany) system equipped with a selective detector with single quadrupole mass (G1956A LC/MSD Quad VL System), a vacuum degassing module (G1379B), a binary pumping module (G1312A), a high performance autosampler (G1367B), a column thermostat (G1316A), and a diode-array detector (G1315A). Data were processed using Agilent LC-MSD ChemStation B.04.03 Data Analysis Software.
An Agilent Zorbax Eclipse Plus Rapid Resolution Stable Bond Poroshell XDB-C18 column 100mm x 4.6mm id and 3.5μm particle size was used for the separation. The analysis was performed as described previously 18 with some modifications. Composition of the mobile phase was: Phase A consisting of 10% (v/v) CH3CN in H2O and 0.05% (v/v) of CH3-COOH; and phase B consisting of 90% (v/v) CH3CN in H2O and 0.05% (v/v) of CH3-COOH. The column elution was carried out with the following linear gradient, 0.0min: 100% phase A with a flow rate of 0.5mL min−1; 39.0min: 100% phase B with a flow rate 0.5mL min−1; 40.0min: 100% phase B with a flow rate of 1.0mL min−1; 41.0min: 100% phase A with a flow rate of 1.0mL min−1; 46.0min: 100% phase A with a flow rate of 1.0mL min−1; 47.0min: 100% phase A with a constant flow rate of 0.5mL min−1. The detector used was of diode arrangement, the temperature of the column at the beginning and end of the test was kept at 25°C and the pressure between 46.8 and 51.3bar, respectively. The injection volume of the sample was 10μL.
Mass analysis was performed by electrospray (ESI-API) using an ionization source of negative polarity. Equipment conditions were: 60 psi nebulizer pressure; drying gas flow of 13 L.min−1; quadrupole temperature at 100°C; gas temperature at 350°C; source current of 8nA at the beginning and 31nA at the end. The ion scan was carried out in full scan mode.
Cytotoxicity and genotoxicity tests. These were performed on the HaCaT cell line (human keratinocytes), established at the Laboratory of Genetic Toxicology, Universidad de Antioquía, Colombia. Cytotoxic effects of the phenolic fractions (PFAc and PFEtOH) were evaluated by two methods, namely a MTT reduction test and trypan blue exclusion test. The genotoxic effect was assessed using the alkaline comet-DNA test.
Cell Cultures: HaCaT cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco BRL) supplemented with 5% of fetal bovine serum (FBS), penicillin (100μg.mL−1), streptomycin (100μg.mL−1) and incubated in a humidified atmosphere of 5% CO2 at 37°C.
MTT reduction test: This is a colorimetric assay for assessing cell metabolic activity and consist in measure of metabolic reduction of 3(4,5-dimethylthiazol-2yl)2,5-diphenyltetrazolium bromide (MTT reagent). The reaction is catalyzed by mitochondrial enzyme succinate dehydrogenase, producing a blue colored compound (formazan). The test determines the mitochondrial functionality of the treated cells, since the amount of living cells is proportional to the amount of formazan produced19.
HaCaT cells were plated at density of 1x104 cells/well in 96-well microplates and maintained under standard culture conditions. After allowing accession, the treatments were performed for each concentration and each sample; after 24 and 48h of incubation 10μL of MTT (Sigma M2128) at 5mg.mL−1 (final concentration in the well: 0.5mg.mL−1) were added and samples were incubated for 4h in the dark. Finally, to dissolve the formazan crystals, 100μL of isopropanol - HCl (0.04M) were added and absorbance at 570nm (A570) was measured using a microplate reader Benchmark Plus (Bio-Rad Laboratories). The percentage of viability was calculated as the following equation.
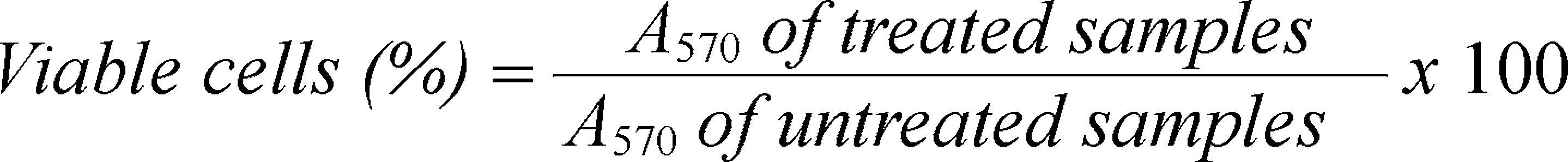
The positive control of the assay was 1mM H2O2 and the negative control was the sample solvent (EtOH). The results are shown as the mean ± SD (standard deviation) of three biological replicates per experimental group.
Trypan blue exclusion test: This test is based on the fact that living cells have intact cell membranes that exclude certain dyes such as trypan blue, while dead cells do not20. HaCat cell were plated (1 x 104 cells/well) into 12-well microplates and were maintained under standard culture conditions. After allowing accession, treatments were performed for each sample and each concentration. After 24 and 48h of exposure, the microplates were processed according to the protocol. For this purpose, cells were detached with a trypsin-EDTA (0.1%) solution and equal volumes of cell suspension and trypan blue 0.4% w/v (Sigma, T6146) were mixed to quantify the cell viability using a hemocytometer. The percentage of living cells was determined by dividing the number total live cells by the total number of cells (both alive and dead cells). For this test, several trials were performed with the purpose of determining the range of concentrations at which cell viability was greater than 80% and thus, based on these resulting ranges, develop the genotoxicity studies. Results are shown as the mean ± SD of three biological replicates per experimental group.
DNA-Comet alkaline test: HaCat cell were plated into 12-well microplates at density of 1 x 104 cells/well and were maintained under standard culture conditions. After allowing accession, treatments were performed for each sample and each concentration under evaluation; after 48h exposure, cells were processed under guidelines of International Workshop on Genotoxicity Test Procedures21, adapted as follows: after treatment, aliquots of 2 x 104 cells were re-suspended in 20μL of simple medium (DMEM) and mixed with 80μL of agarose low melting point (Sigma, A9414 molecular biology grade) at 37°C. Afterwards, the cell/agarose mix was layered on glass microscope slides, pre-coated with agarose of normal fusion point (Sigma, A9539 molecular biology grade). The coated slides were kept at 4°C for 15min to allow solidification of the agarose. Subsequently, cells were then subjected to digestion for 1 hour in lysis buffer (2.5M NaCl, 0.1M Na2EDTA, 10mM Tris base, 1% N-lauryl sarcosinate, 10% DMSO and 1% Triton X-100 (Sigma, T8787), adjusted at pH 10.0) and 30min in denaturation buffer (50mM NaOH/1mM EDTA, pH 13). After, slides were place in a horizontal submarine gel electrophoresis system (Bio-Rad Co., Hercules, CA, USA) containing Tris-Borate-EDTA buffer. The electrophoresis conditions were 25 volts and 300mA. Then, plates were washed three times for 5min with neutralizing buffer pH 7.4 (0.4M Tris). Finally, cells were dyed with 40μL of ethidium bromide (20μg.mL−1) (Sigma, E7637) and plates were read in a fluorescent microscope (Leica DMLS, Austria) under magnification 20X, excitation filter 515-560nm and barrier filter 590nm. For each treatment, 50 cells were photographed and subsequently analyzed with the Comet Assay Software Project (Casp, http://casp.sourceforge.net) for quantifying percentage of DNA in the queue. According to the amount DNA in the tail, nuclei were classified into one of following categories: a) no damage DNA, b) low damage, c) moderate damage d) high damage. The positive control in this trail was 0.3mM hydrogen peroxide (30min exposure) and the negative control was untreated cells.
Statistical analysis. The results are presented as the value of the mean ± standard deviation (SD). Statistical analyses were performed using Statistical Package for Social Sciences (SPSS) software (SPSS Inc., Chicago, IL, USA). For quantifying total phenolic content, five independent replicates were performed. One-way analysis of variance (ANOVA) with Tukey's HSD (Honestly Significant Difference) post-hoc test was used to determine the possible differences among treatment. The cytotoxicity and genotoxicity tests were performed in triplicate for each experimental group and data were analyzed using the Bonferroni's multiple comparison tests, the mean difference (mean.dif) and the confidence interval at 95% of mean.dif were calculated. P values < 0.05 were considered to be statistically significant.
RESULTS AND DISCUSSIONExtracts and total phenolic content. The color of the extracts obtained was between yellow to reddish. The ethyl acetate extracts were the ones with the lighter shade, while the darkest were the acetone extracts. Fractions PFAc and PFEtOH had the highest content of phenols (PFAc = 11.34 ± 0.88 mgGAE/g; PFEtOH = 6.52 ± 1.28 mgGAE/g; PFBuOH = 0.713 ± 0.23 mgGAE/g; PFEtAc = 0.15 ± 0.17 mgGAE/g). ANOVA shows that the treatments means aren’t all equal (p value = 7.18 x 10−11). In addition, pairwise comparison (Tukey's HSD test) showed differences between all treatments (p value < 0.001), except for PFEtAc - PFBuOH couple (mean.dif: -0.562; p value: 0.679; IC95: -2.042 to 0.917).
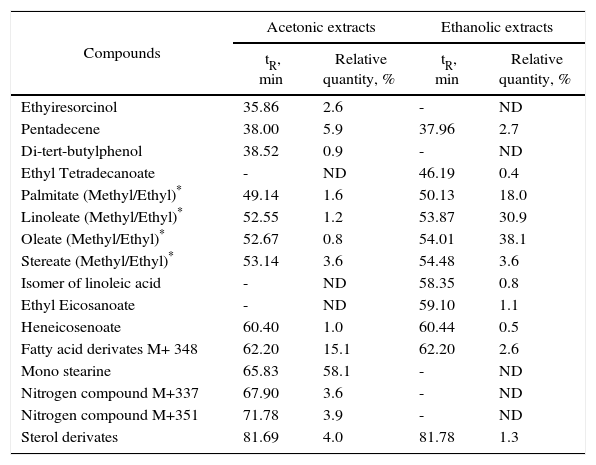
GC-MS and HPLC-MS. GC-MS analysis identified two phenolic compounds in the acetone extracts, these are: ethyl-resorcinol and di-tert-butylphenol, with a relative abundance of 2.6% and 0.9% respectively. Other compounds present in ethanolic and acetonic extracts were: pentadecene, esters of saturated and unsaturated fatty acids, fatty acid derivatives and sterol derivatives (Table I).
Tentative identification and relative abundance (%) of compounds in acetonic and ethanolic extracts from Ulomoides dermestoides beetles, determined by gas chromatography coupled to mass spectrometry (GC-MS).
| Compounds | Acetonic extracts | Ethanolic extracts | ||
|---|---|---|---|---|
| tR, min | Relative quantity, % | tR, min | Relative quantity, % | |
| Ethyiresorcinol | 35.86 | 2.6 | - | ND |
| Pentadecene | 38.00 | 5.9 | 37.96 | 2.7 |
| Di-tert-butylphenol | 38.52 | 0.9 | - | ND |
| Ethyl Tetradecanoate | - | ND | 46.19 | 0.4 |
| Palmitate (Methyl/Ethyl)* | 49.14 | 1.6 | 50.13 | 18.0 |
| Linoleate (Methyl/Ethyl)* | 52.55 | 1.2 | 53.87 | 30.9 |
| Oleate (Methyl/Ethyl)* | 52.67 | 0.8 | 54.01 | 38.1 |
| Stereate (Methyl/Ethyl)* | 53.14 | 3.6 | 54.48 | 3.6 |
| Isomer of linoleic acid | - | ND | 58.35 | 0.8 |
| Ethyl Eicosanoate | - | ND | 59.10 | 1.1 |
| Heneicosenoate | 60.40 | 1.0 | 60.44 | 0.5 |
| Fatty acid derivates M+ 348 | 62.20 | 15.1 | 62.20 | 2.6 |
| Mono stearine | 65.83 | 58.1 | - | ND |
| Nitrogen compound M+337 | 67.90 | 3.6 | - | ND |
| Nitrogen compound M+351 | 71.78 | 3.9 | - | ND |
| Sterol derivates | 81.69 | 4.0 | 81.78 | 1.3 |
tR = Retention time.
ND = no detected.
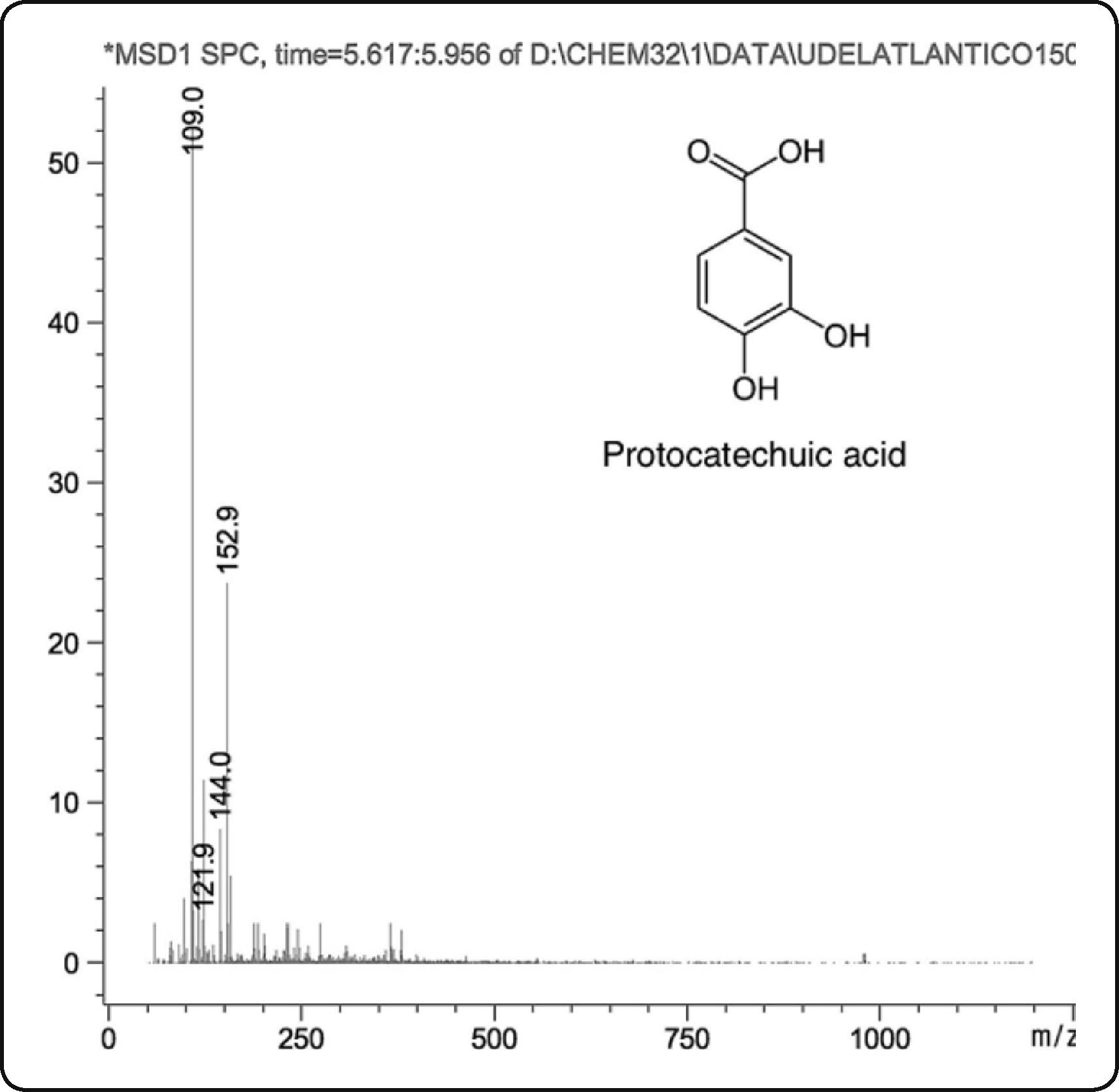
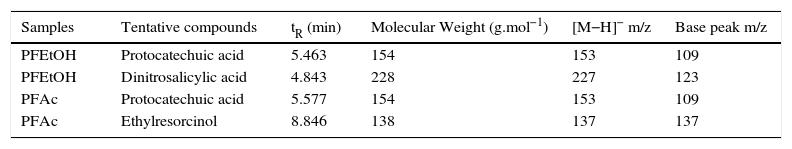
By HPLC-MS, the following compounds were tentatively identified: 3,4-dihydroxybenzoic or protocatechuic acid, dinitrosalicylic acid and ethylresorcinol (Table II). Protocatechuic acid (abbreviation PCA) was common in both fractions (Figure 1); this compound was found in the cuticle of arthropods, such as cockroaches (Blatella orientalis and Periplaneta americana)22–24 and beetles (Pachynoda epphipiata and Tenebrio molitor)25. PCA seems to play the important role in obscuring the cuticle and capturing free radicals. This compound is widely distributed in plant kingdom22, being extracted by their pharmacological properties as antioxidant agent26,27.
Relative chemical composition of phenolic fractions of ethanolic extracts (PFEtOH) and acetonic extracts (PFAc) of Ulomoides dermestoides, determined by HPLC-MS-ESI in negative ion mode.
| Samples | Tentative compounds | tR (min) | Molecular Weight (g.mol−1) | [M−H]− m/z | Base peak m/z |
|---|---|---|---|---|---|
| PFEtOH | Protocatechuic acid | 5.463 | 154 | 153 | 109 |
| PFEtOH | Dinitrosalicylic acid | 4.843 | 228 | 227 | 123 |
| PFAc | Protocatechuic acid | 5.577 | 154 | 153 | 109 |
| PFAc | Ethylresorcinol | 8.846 | 138 | 137 | 137 |
tR = Retention time. All compounds were tentatively identified according to their pseudomolecular ion, therefore the chemical identity must be verified with standard compounds.
![Mass spectrum of the protocatechuic acid in ethanolic and acetonic extracts of Ulomoides dermestoides. Observe the molecular ion [M−H]− at m/z 153 atomic mass unit (amu) and base peak [M−H−CO2]− at m/z 109 amu.](https://static.elsevier.es/multimedia/1405888X/0000001900000002/v1_201607220355/S1405888X16300018/v1_201607220355/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcNGSBFqzjIokZJFmIUKi41jgeDgOhiQKNqiaP/upQ0VSPuJGeQ9nhouKQfAHU/SrRkuxq27CCOy4c80OcL4yp5KT783htno7Hl1uiTrKfSsoP9tQhw3vtkJT2aqs+vGrgglETomgQ1qeP6ZcDspbsqukCmgpLnhoLKP4TpaZLy3Dnacc8zwO+zLhPFaGESYJO7RatuAnE6W5u1Yt8WCOliwjmsKAWzGCeQI1EOe3pPRw== "Mass spectrum of the protocatechuic acid in ethanolic and acetonic extracts of Ulomoides dermestoides. Observe the molecular ion [M−H]− at m/z 153 atomic mass unit (amu) and base peak [M−H−CO2]− at m/z 109 amu.")
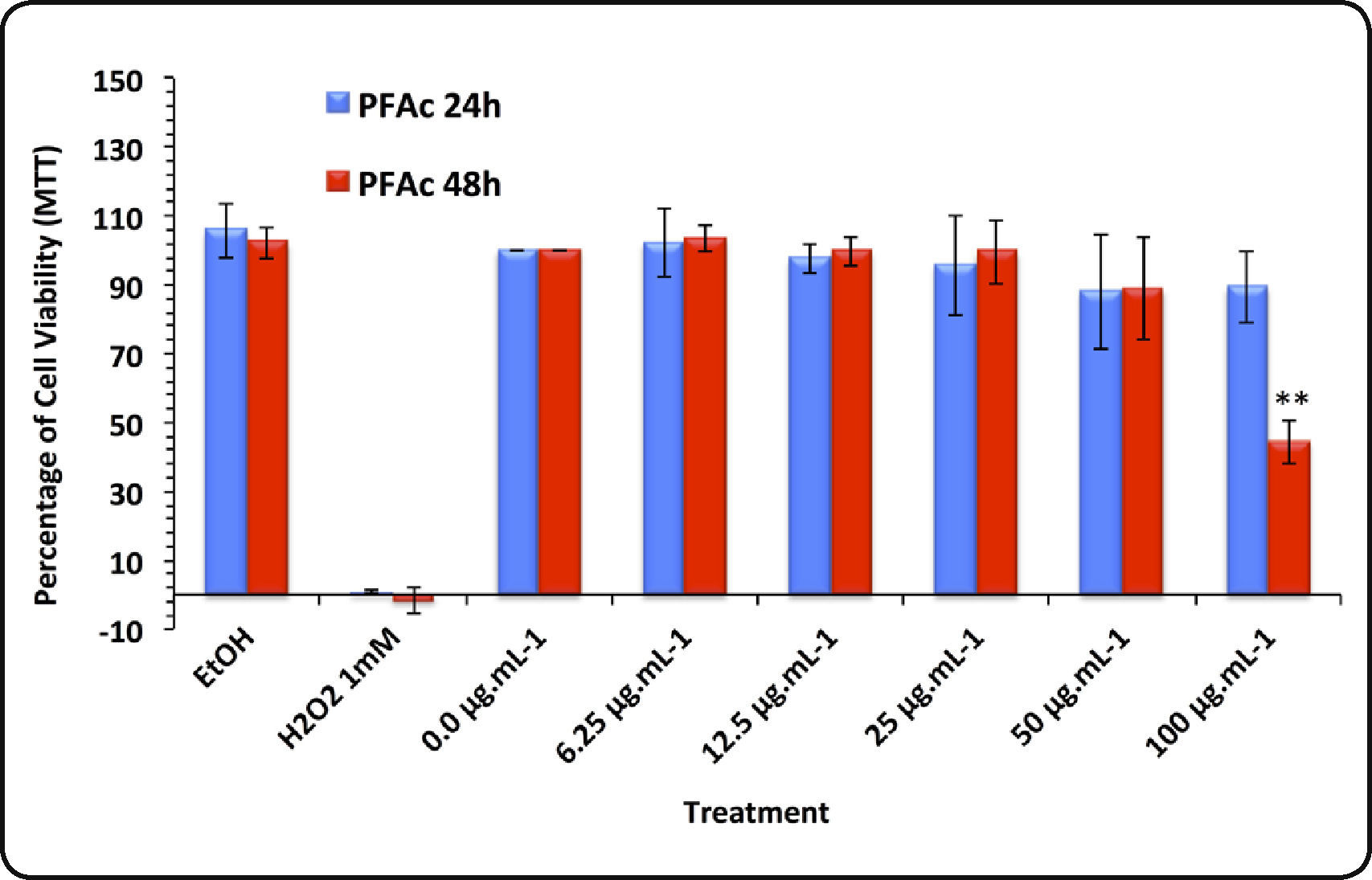
Cytotoxicity tests. Results of MTT reduction test using HaCat cells are presented in Figure 2. None of the treatments with PFAc showed significant decrease of cell viability at 24h exposure. At 48h, the cell viability decreased by 44.2 ± 6.2% in the treatment with 100μg.mL−1 (IC50 = 91.85 ± 12.05mg.mL−1) (Figure 2A).
. Results are given as mean ± SD of assays performed in triplicate. Asterisk indicates statistically significant differences (**p value < 0.01) with respect to untreated cells.")
Results of the MTT assay conducted in HaCat cells treated with phenolic fractions of acetonic extracts of Ulomoides dermestoides beetles. Pair of bars represents assays with different times of exposition to the treatments (24 and 48hours). Results are given as mean ± SD of assays performed in triplicate. Asterisk indicates statistically significant differences (**p value < 0.01) with respect to untreated cells.
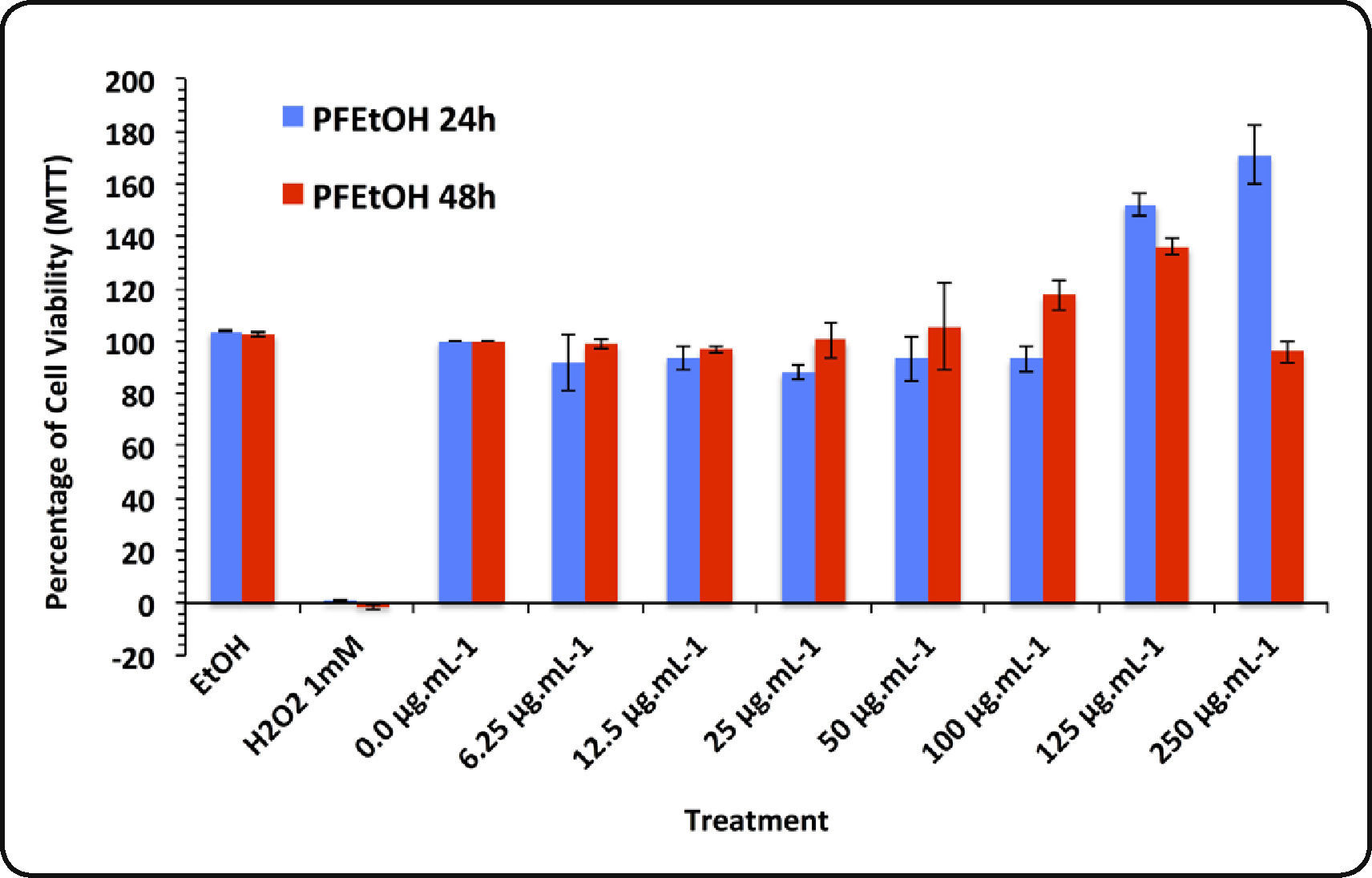
Moreover, HaCat cells treated with PFEtOH showed survival higher than 100%, at concentrations > 100μg mL−1 (Figure 2B). A cell-free assay showed that PFEtOH react with MTT reagent (data not shown). This behavior has been reported with certain plant extracts28 and with redox-active polyphenolic compounds, which may directly reduce tetrazolium salts, even without cells29–31. Although the MTT assay is widely used to measure the cellular proliferation, cell viability and drug toxicity, several studies report compounds that increase MTT reduction with no enhance of cell viability30,32, a fact that should be considered when natural extracts are evaluated, to avoid false results. For this, we propose that MTT reduction test is not a reliable method to assess the cytotoxicity of extracts from U. dermestoides.
. Results are given as mean ± SD of assays performed in triplicate.")
Results of the MTT assay conducted in HaCat cells treated with phenolic fractions of ethanolic extracts of Ulomoides dermestoides beetles. Pair of bars represents assays with different times of exposition to the treatments (24 and 48hours). Results are given as mean ± SD of assays performed in triplicate.
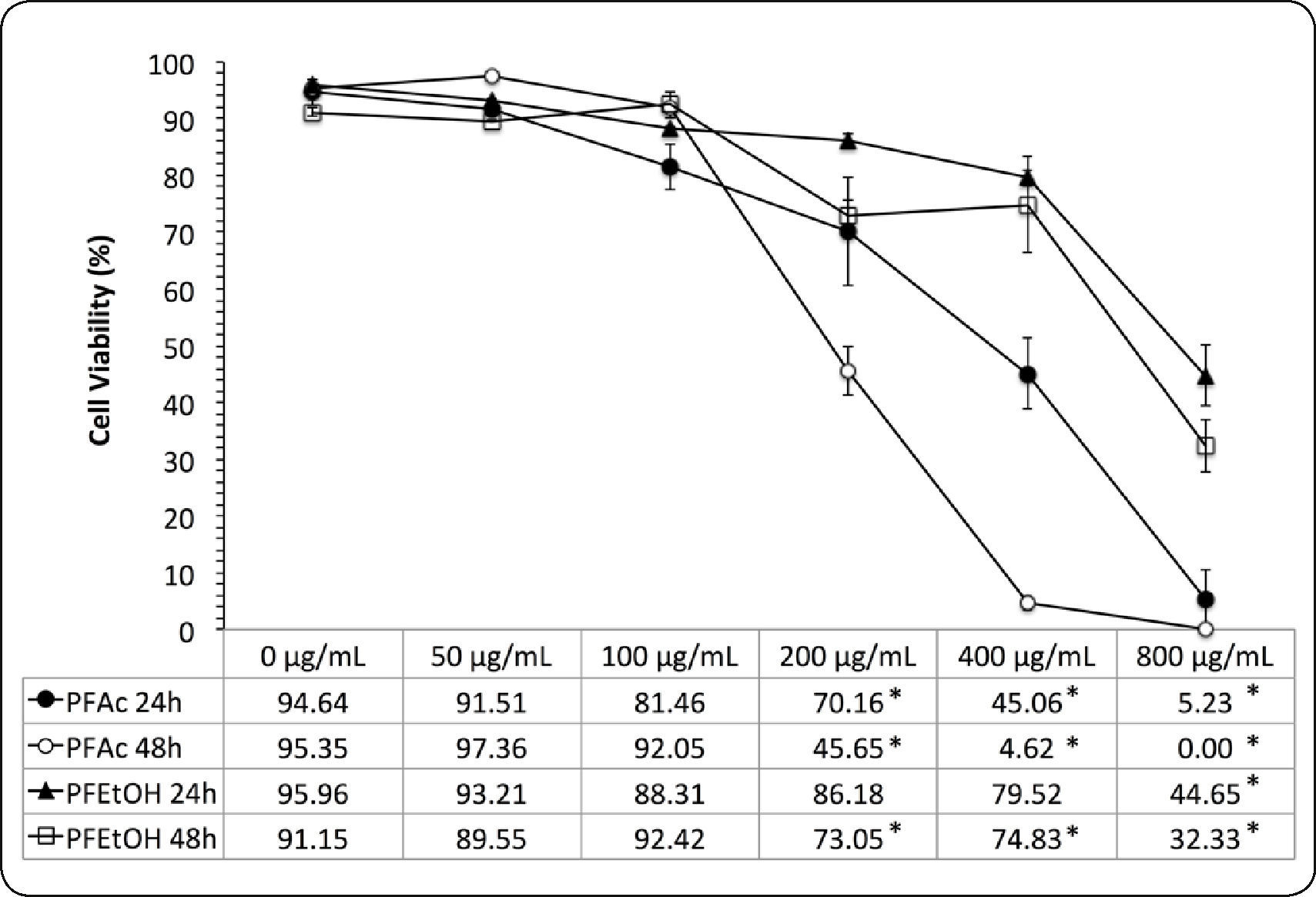
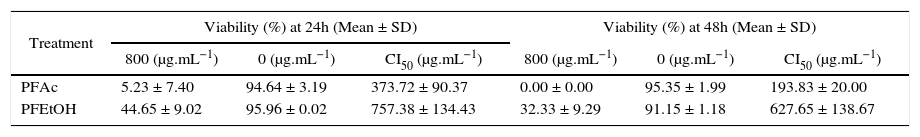
Results with the trypan blue exclusion test show that viability of HaCaT cells is dependent on the concentration and time of exposure to each treatment (Figure 3). Cell viability, at 24 and 48h was higher in treatments with PFEtOH (CI50 of 757.39 ± 134.43μg.mL−1 and 627.65 ± 138.67μg.mL−1, respectively). In treatments with PFAc, CI50 were 373.72 ± 90.37μg.mL−1 and 193.83 ± 20.00μg.mL−1, respectively (Table III).
. The viability of HaCaT cells is reported in function of concentration of samples (0 to 800μg.mL−1). Tests were performed for 24 and 48hours. Asterisk indicates statistically significant differences (*p value < 0.05) with respect to untreated cells. The mean ± SD is shown.")
Dose-response curves of phenolic fractions of Ulomoides dermestoides (Trypan blue exclusion test). The viability of HaCaT cells is reported in function of concentration of samples (0 to 800μg.mL−1). Tests were performed for 24 and 48hours. Asterisk indicates statistically significant differences (*p value < 0.05) with respect to untreated cells. The mean ± SD is shown.
Results of the trypan blue exclusion test in HaCaT cells exposed to different concentrations of phenolic fractions of Ulomoides dermestoides extracts.
| Treatment | Viability (%) at 24h (Mean ± SD) | Viability (%) at 48h (Mean ± SD) | ||||
|---|---|---|---|---|---|---|
| 800 (μg.mL−1) | 0 (μg.mL−1) | CI50 (μg.mL−1) | 800 (μg.mL−1) | 0 (μg.mL−1) | CI50 (μg.mL−1) | |
| PFAc | 5.23 ± 7.40 | 94.64 ± 3.19 | 373.72 ± 90.37 | 0.00 ± 0.00 | 95.35 ± 1.99 | 193.83 ± 20.00 |
| PFEtOH | 44.65 ± 9.02 | 95.96 ± 0.02 | 757.38 ± 134.43 | 32.33 ± 9.29 | 91.15 ± 1.18 | 627.65 ± 138.67 |
PFAc = Phenolic fraction of acetonic extract.
PFEtOH = Phenolic fractions of ethanolic extract.
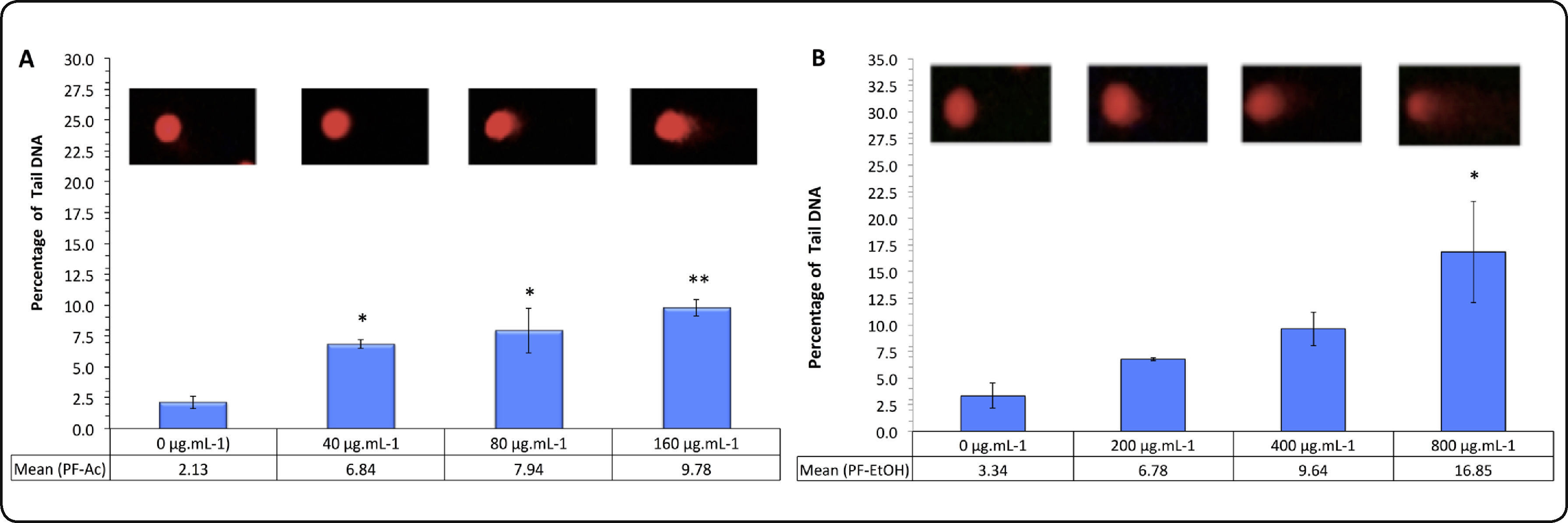
Genotoxicity tests. Results of DNA-comet alkaline test, showed statistically significant difference (p < 0.05) between the untreated and treated groups with different concentrations of PFAc, with intermediate genotoxic effect to 160μg.mL−1 (Mean.dif: -7.646; IC95: -11.65 to -3.641) (Figure 4). On the other hand, treatment with PFEtOH resulted in a low genotoxic effect, at concentrations equal to or less than 400μg.mL−1; statistically significant difference between the untreated and treated groups were observed at concentrations ≥ 800μg.mL−1 (Mean.dif: -13.52; IC95: -23.71 to -3.323). The positive control of the trial (0.3mM H2O2) produced cores with high damage (% DNA in the tail: 57.55 ± 0.34%); comparatively, the DNA damage in cells exposed to solvent EtOH (negative control) was very low (% DNA in the tail: 2.734 ± 1.02%).
 acetonic extracts of Ulomoides dermestoides and (B) ethanolic extracts of Ulomoides dermestoides. The top panel shows the comets for each treatment after 24h. Values are expressed as means of percentage of DNA in the comet tail ± SD. Statistical comparisons were made using one-way ANOVA/Bonferroni post-hoc test (*p value <0.05; **p value <0.01 with respect to untreated cells).")
DNA damaged in alkaline comet assay using HaCaT cells after 24 h incubations with phenolic fractions of (A) acetonic extracts of Ulomoides dermestoides and (B) ethanolic extracts of Ulomoides dermestoides. The top panel shows the comets for each treatment after 24h. Values are expressed as means of percentage of DNA in the comet tail ± SD. Statistical comparisons were made using one-way ANOVA/Bonferroni post-hoc test (*p value <0.05; **p value <0.01 with respect to untreated cells).
We suggested that the higher cytotoxic and genotoxic activity of PFAc could be related to the higher content of phenolic compounds, as PCA. Several studies report the antioxidant and free radical capturing activity of PCA33–36; however, this compound has also been reported as a pro-oxidant, depending on their concentration37. It has even been shown that in high doses PCA could induce lipid peroxidation dependent on the Fe2+/Fe3+ redox cycle and oxidative DNA damage dependent on the Cu1+/Cu2+ redox cycle38,39.
A study by Babich et al., 2002 describes oxidative stress inducing in culture of non-tumorigenic gingival epithelial cells (SG) and cultured malignant cells derived from salivary glands (HSG1), exposed to > 10mM PCA. These authors suggest a mechanism of PCA-mediated activation of tyrosinase enzymes leading to production of an active metabolite (quinone) capable of oxidizing the glutathione (GSH), altering cellular defense systems against ROS40. On the other hand, Galati et al., 2002, proposed that phenolic rings of certain polyphenols could be metabolized by peroxidases to form pro-oxidant phenolic radicals, reactive enough to co-oxidize the GSH and the NADH coenzyme, with the sub-sequent formation ROS41.
It should also be considered that, under certain experimental conditions, some phenolic compounds could initiate autoxidation reactions, behaving as pro-oxidant molecules in culture media such as DMEM and RPMI42. For example, it has been found that in the presence of DMEM, high doses of (-)-epigallocatechin-3-gallate, (+)-catechin and quercetin produce toxic levels of H2O2 (frequently > 50μM), semiquinone and quinones43–46. In addition, high doses of phenolic acids were also associated with DNA damage induced by H2O247. It has been described that exposure during 6 hour, to doses of up to 50 mmol L−1 of purified flavonoids, could cause chromosomal translocation and clastogenicity in human cell lines48,49.
Given that our experiments were performed in cells cultured in DMEM, it is possible that the decrease in cell viability is related to H2O2 production from auto-oxidation of phenolic compounds present in extracts of U. dermestoides analyzed.
CONCLUSIONSEthanolic and acetonic extracts from whole body of Ulomoides dermestoides are a source of redox-reactive phenolic compounds that could be of biomedical interest. In both extracts was tentatively identified the protocatechuic acid, but further analysis with the standard compound would be required to confirm this assignment. Additionally, experimental design developed in this study suggests that the beetle's ethanol extracts shown low toxicity in HaCaT cell line, at the tested concentrations. We suggest that the greater cytotoxicity/genotoxicity of acetonic extracts could be related to a higher concentration of redox-reactive compounds present in the sample and the time of exposure to these compounds.