


Introducción
El mecanismo de acción del gamma hidroxibutirato (GHB) no está del todo dilucidado. La droga fue sintetizada por primera vez por Laborit1 en busca de un análogo del ácido gamma aminobutírico (GABA) capaz de penetrar la barrera hematoencefálica. La sustitución de un hidroxilo en posición de un grupo amino confirió esta propiedad a la nueva molécula, aunque sus efectos en animales de experimentación resultaron ser diferentes de aquéllos producidos por GABA tanto a nivel bioquímico como a nivel conductual.
GHB y el sistema GABAérgico
La hipótesis sugiriendo el sistema GABAérgico como mecanismo de acción del GHB está basada en numerosos estudios. En primer lugar se ha descrito cierta afinidad de GHB por el receptor GABAB2, actuando como agonista parcial, pero no por el receptor GABAA3, si bien esta afinidad es a concentraciones superiores a aquéllas presentes normalmente en el cerebro. Por otra parte, se ha sugerido que GHB podría estar actuando como precursor de GABA debido a la existencia de una enzima capaz de oxidar GHB a semialdehído succínico (SSA) (GHB deshidrogenasa) y ésta ser reconvertida a GABA2,4,5, ya que la administración de un precursor de GHB, la gammabutirolactona (GBL), produce un efecto parecido al agonismo GABAérgico6.
Por otra parte, en ratones GABAB -/-, GHB no produce hipolocomoción, hipotermia ni un incremento en la síntesis de dopamina ni incrementos en las ondas delta del encefalograma, efectos observados en los ratones wild-type7. Estas evidencias parecen indicar que al menos algunos de los efectos producidos por GHB implican a los receptores GABAB.
Sin embargo, GHB no produce constantemente el mismo perfil de efectos que un agonista GABA, indicando que existiría al menos otro mecanismo de acción del GHB.
El GHB endógeno se ha propuesto como un neurotransmisor o neuromodulador, ya que cumple muchos de los criterios necesarios para ser considerado como tal. Existe una distribución anatómica discreta y subcelular de GHB y de su enzima de síntesis8 en terminales presinápticos en el cerebro. Las concentraciones más altas de GHB en el cerebro se encuentran en la sustancia negra9 y el hipotálamo4. GHB es liberado de la neurona de manera calcio-dependiente por despolarización inducida por potasio y existe un sistema de captación de alta afinidad dependiente de sodio10,11, siendo el estriado al área cerebral con el índice de captación más alto11.
Por otra parte, existen lugares específicos de fijación de [3H]GHB en el cerebro de rata12 que son de alta afinidad cuya distribución se correlaciona con el recambio de GHB y que parecen estar colocalizados en estructuras dopaminérgicas13. Recientemente se han clonado estos supuestos receptores GHB en rata14 y en tejido humano15. Los receptores están acoplados a proteínas G, pero no muestran homología de secuencia con otros receptores de la familia de receptores acoplados a proteínas G (GPCR) que incluyen el receptor GABAB. Tanto el receptor clonado en rata como aquél clonado en tejido humano son lugares de alta afinidad para la fijación de GHB, pero comparten poca homología entre ellos y mientras que el receptor clonado en rata parece estar asociado a un canal mixto catiónico (Na+/K+) y no muestra fijación del antagonista NCS-382 (ácido [2E]-[5-hidroxi-5,7,8,9-tetrahidro-6H-bezo(a)(7)anulen-6-ilidene]etanoico)14 aquél clonado en el cerebro humano está asociado a un canal más selectivo para Na+ y posiblemente para Ca2+ y además sí muestra fijación de NCS-38215. Sin embargo, la localización celular de los receptores GHB no está del todo dilucidada. Estudios que han utilizado animales inyectados directamente en estriado con ácido kaínico o con lesiones dopaminérgicas inducidas por 6-hidroxidopamina (6-OHDA) han demostrado que los receptores GHB no están presentes en los terminales presinápticos de dopamina sino en neuronas intrínsicas16 que podrían ser GABAérgicas y/o encefalinérgicas. Por tanto, la liberación local de GABA o encefalinas podría estar mediada por estos receptores.
El control de la liberación de GABA mediante estos receptores explicaría, por tanto, los efectos inducidos por GHB pero mediados por GABAB y la prevención por naloxona tanto de la disminución en la liberación de dopamina6 como del incremento en la síntesis de dopamina en el estriado17.
GHB y el sistema dopaminérgico
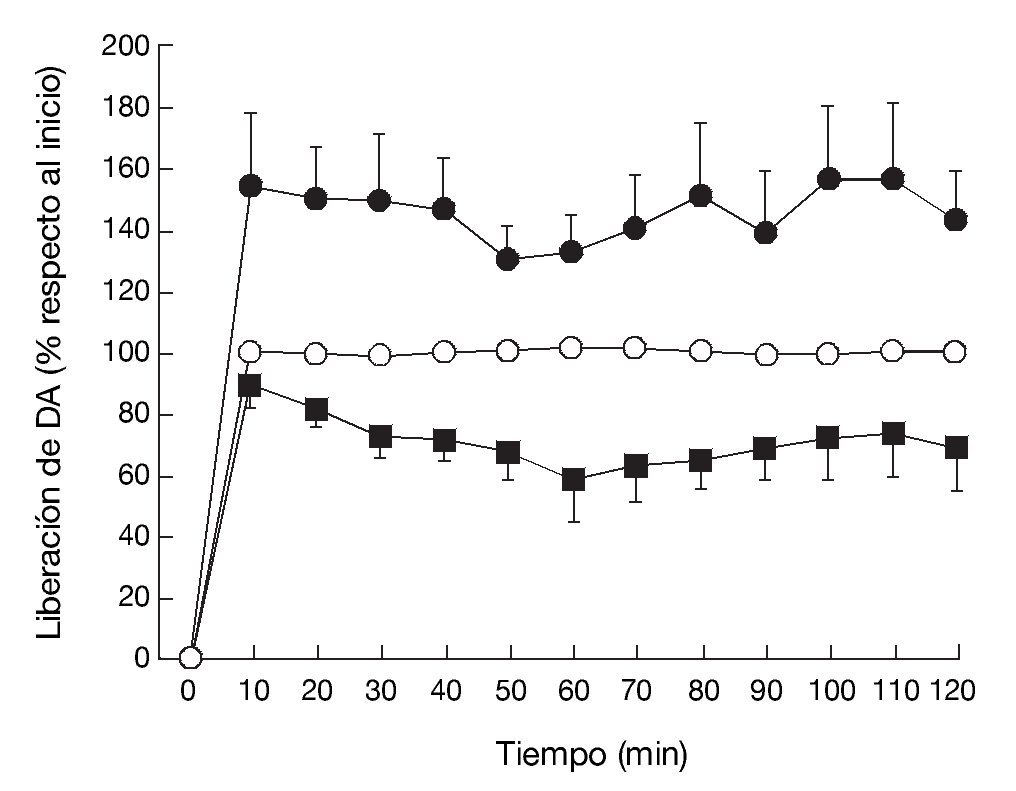
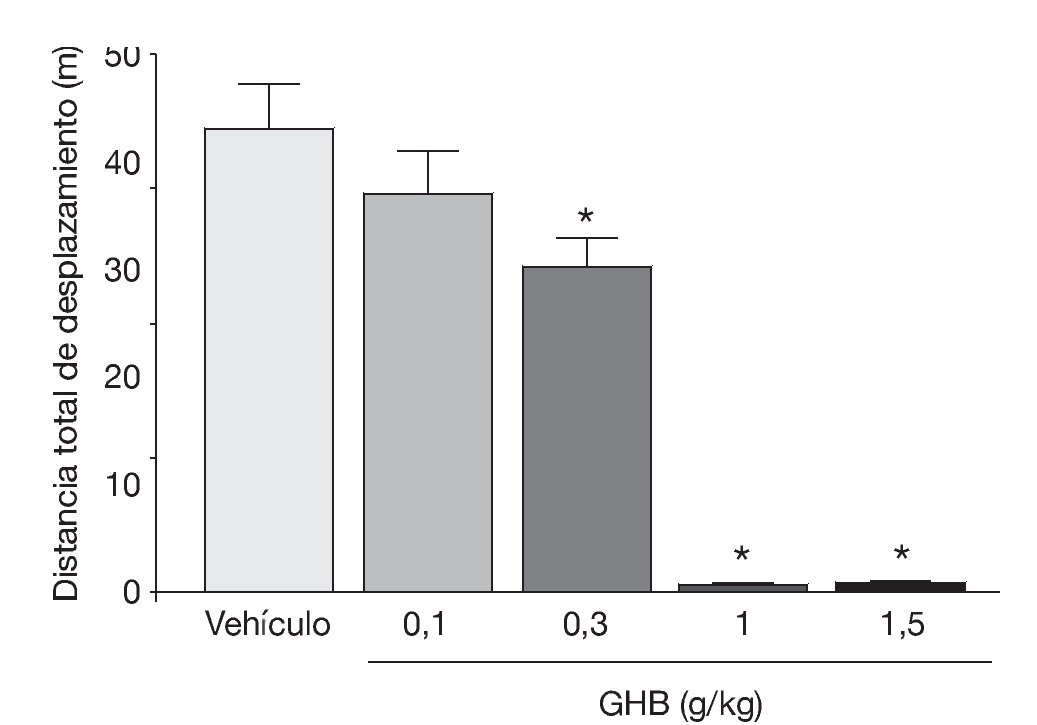
La administración exógena de GHB produce una modulación de varios sistemas de neurotransmisión, entre ellos el dopaminérgico, el cual se cree responsable de algunos de los efectos motores y conductuales del GHB. Mediante estudios de microdiálisis in vivo en animal despierto se ha demostrado que GHB produce una inhibición de la liberación de dopamina (fig. 118). De acuerdo con esta observación, estudios en roedores han demostrado sedación y una disminución dosis-dependiente de la actividad locomotora (fig. 2)7,19 tras la administración de GHB. Además, GHB reduce el comportamiento estereotipado y la hipotermia producidos por la hiperestimulación de los receptores dopaminérgicos inducida por apomorfina19,20, probablemente mediante una reducción en el efecto contributivo de la dopamina liberada de forma espontánea5. Esta reducción en la liberación de dopamina es inhibida por antagonistas del receptor GABAB, implicando a este receptor en el efecto dopaminérgico21.
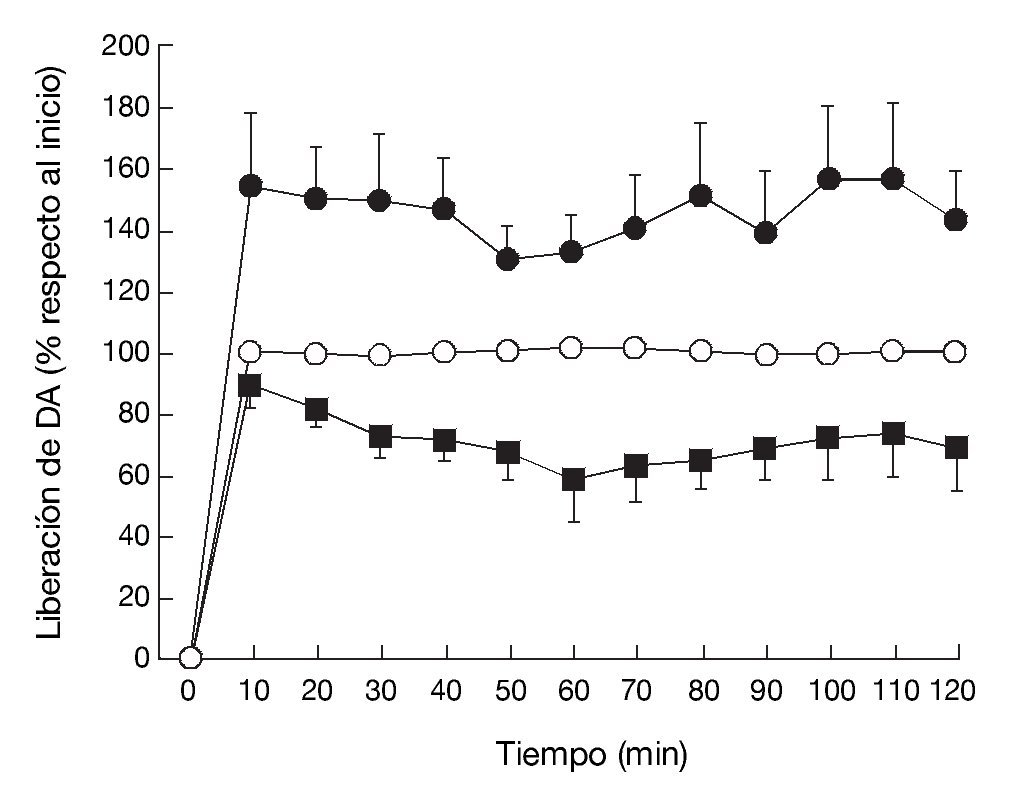
Figura 1. Efecto de (*) vehículo, (•) GHB (500 mg/kg), i.p.) o (*) GHB + naloxona (0,8 mg/kg, i.p.) sobre los niveles de dopamina en el dializado estriatal en rata despierta.
Tomada de Feigenbaum JJ et al18.
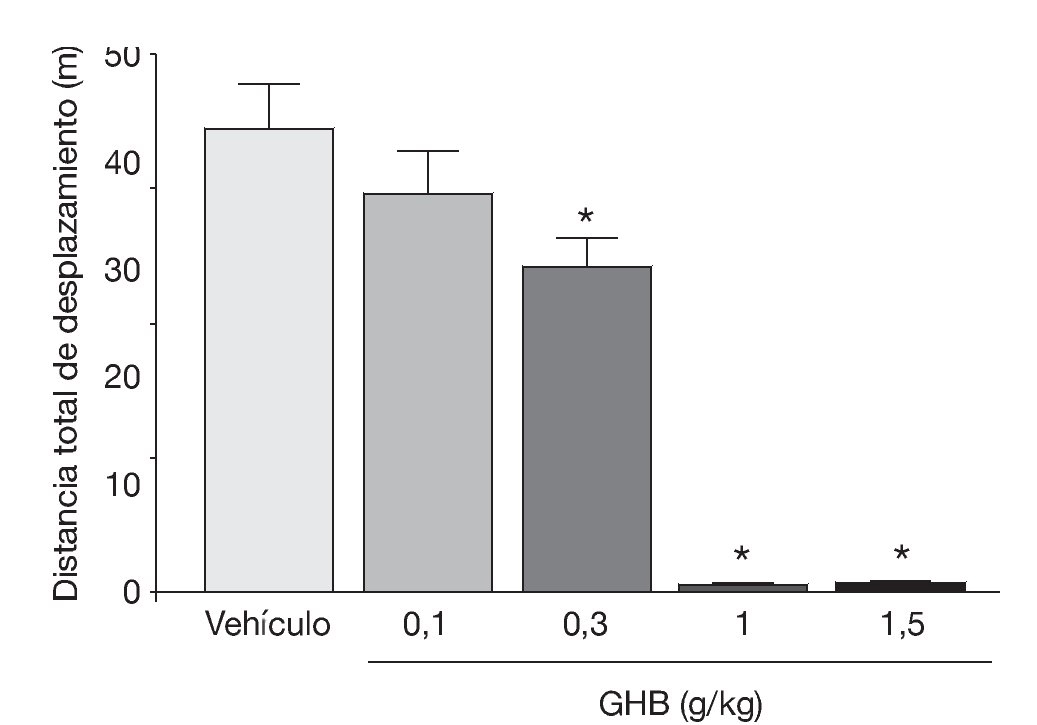
Figura 2. Disminución dosis-dependiente en la actividad motora en ratones BALB/c producida por GHB. *p<0,05 frente a animales tratados con vehículo (test de Dunnett).
Tomada de Kaupmann K et al7.
En dosis altas, la rápida disminución en la liberación de dopamina es seguida por una acumulación de dopamina en tejido del cerebro frontal22 debido seguramente a un incremento en la actividad de la tirosina hidroxilasa (enzima limitante de la síntesis de dopamina), ya que este incremento de dopamina es bloqueado por la administración de a-metiltirosina23. La acumulación de dopamina es seguida por un incremento en la liberación de la misma en el estriado y áreas de la vía dopaminérgica corticolímbica24. Este incremento en la liberación de dopamina es rápido a dosis altas de GHB o si la administración de la droga es mediante infusión directa en el estriado, pudiendo llegar a encubrir el efecto inicial de disminución de la liberación de dopamina y, por tanto, dando lugar a observaciones conflictivas. La administración del antagonista del receptor GHB, NCS-382, previene el incremento en la concentración extra-celular de dopamina inducido por GHB5, implicando a los receptores GHB en este efecto dopaminérgico.
Por otra parte, el uso de animales anestesiados en vez de despiertos o la utilización de diferentes vías de administración (por ejemplo, intraperitoneal frente a subcutánea) podrían estar contribuyendo a las diferencias en respuestas dopaminérgicas observadas25. Además, es posible que los efectos de la droga sobre la liberación de dopamina sean diferentes en distintas áreas cerebrales por la distribución de los diferentes receptores implicados en el efecto y la sensibilidad de los mismos.
GHB y el sistema opioide
La administración local de GHB produce un incremento en la liberación de opioides endógenos, si bien esta liberación de opioides parece estar mediada indirectamente por la disminución de la función dopaminérgica26. Estudios realizados utilizando GBL (precursor de GHB que produce un incremento en las concentraciones cerebrales de éste), indican que la naloxona es capaz de atenuar o bloquear las alteraciones en el electroencefalograma producidos por el precursor, así como el incremento en dopamina estriatal producido por el mismo. Dosis parecidas de GBL producen un aumento en la concentración de dinorfina A en el hipocampo y la glándula pituitaria y una disminución del contenido de b-endorfina en el tálamo, el hipotálamo y la glándula pituitaria27. In vitro la aplicación de antagonistas de opiáceos a rebanadas de hipocampo previene el incremento en el recambio de cGMP y de inositol fosfato inducidos por GHB28. Por otra parte, se ha propuesto que gran parte de los efectos conductuales, cambios en el electroencefalograma y en dopamina podrían ser reproducidos por morfina, encefalinas y b-endorfinas. Sin embargo, GHB no se une a los receptores mu, delta y kappa26, por lo cual se puede descartar un efecto directo sobre estos receptores.
GHB y el sistema serotonérgico
Dosis farmacológicas de GHB (400-500 mg/kg) producen un incremento en el recambio de 5-HT en el estriado y en áreas mesolímbicas29. El contenido de serotonina no se ve modificado, aunque la síntesis de 5-HT se incrementa evidenciada por la acumulación de 5-hidroxitriptófano (producto intermediario en la síntesis de 5-HT) tras la inhibición de L-aminoácido aromático descarboxilasa. Además, el principal metabolito de 5-HT, el ácido 5-hidroxiindol ácetico (5-HIAA) se acumula en las mismas áreas cerebrales5. Sin embargo, y a pesar del incremento en el recambio de serotonina, GHB no potencia el aumento en los niveles extracelulares de 5-HT inducido por potasio30.
GHB produce el incremento en la síntesis de 5-HT mediante un aumento en la biodisponibilidad de triptófano y no mediante una modificación en las características cinéticas de la triptófano hidroxilasa (enzima limitante de la síntesis de 5-HT5). Una acumulación de triptófano en el cerebro de animales tratados con GHB (posiblemente mediante un incremento en el transporte a través de la barrera hematoencefálica o membranas neuronales) podría ser responsable del incremento en la síntesis de 5-HT.
Queda aún por dilucidar la participación de diferentes receptores en este efecto de GHB. La administración de un agonista (NCS-356) o un antagonista (NCS-382) de los receptores GHB reproduce o previene, respectivamente, la acumulación de triptófano y de 5-HIAA. Por otra parte, algunos de los efectos de GHB pueden ser reproducidos por baclofén, un agonista GABAB, y antagonizados por CG35348 (ácido [3-amino-propil][dietoximetil]fosfínico), un antagonista específico de este mismo receptor. Todo esto indica que los efectos de GHB sobre el sistema serotonérgico podrían estar mediados por la acción presináptica de los receptores GHB sobre la liberación de GABA5.
GHB y adicción
Con el fin de evaluar similitudes y diferencias entre GHB y otras drogas de abuso se han realizado estudios de discriminación. Se ha sugerido que este tipo de estudio en animales apunta a dianas celulares comunes entre drogas que se sustituyen unas a otras. Además, parecen ser útiles en la evaluación del potencial de abuso de drogas. En un estudio realizado por Winter et al31 utilizando una serie de compuestos en animales entrenados para discriminar GHB de salino, únicamente se observó sustitución parcial con morfina, dietilamida del ácido lisérgico (LSD), d-anfetamina, agonistas GABA y clordiazepóxido. Barbital y compuestos relacionados con fenciclidina (PCP) no fueron capaces de sustituir a GHB.
Sin embargo, baclofén, un agonista en los receptores GABAB, sustituyó a GHB y en especial en dosis altas de GHB. El antagonista de receptores GABAB, CGP35348, bloqueó la discriminación de dosis altas y bajas de GHB, pero fue más eficaz en dosis altas de GHB32. Esto indica que a dosis bajas el efecto discriminatorio de GHB está mediado por los receptores GHB, mientras que en dosis altas predomina un efecto sobre los receptores GABAB33.
Etanol ha demostrado sustituir parcialmente a GHB31, pero este efecto parece observarse únicamente en dosis intermedias de etanol (1.000 mg/kg, i.g.), desapareciendo tanto en dosis más altas como en dosis más bajas34.
Esta misma dosis de etanol es sustituida por GHB.
Estudios en monos han demostrado una sustitución parcial dosis-independiente de GHB en animales entrenados para discriminar anfetamina, pero ningún efecto en animales entrenados para discriminar pentobarbital35.
En estudios de refuerzo GHB produjo una preferencia de plaza36, pero únicamente después de 6 exposiciones a la droga sugiriendo un refuerzo más débil que aquél producido por otras drogas de abuso como cocaína u opiáceos que requieren menos exposiciones.
Los estudios de autoadministración son controvertidos. En un estudio sobre el consumo de GHB en agua de bebida, ratas criadas selectivamente para autoadministrar etanol aprendieron a beber GHB con más facilidad que otras cepas34,37. Tras un período de dos semanas con GHB en el agua de bebida (1% p/v) los animales fueron expuestos a dos bebederos: uno con agua y el otro con GHB (1%). Las ratas bebieron más GHB que agua de forma alternativa en ciclos de 1-2 días. La razón de este patrón alternativo de preferencia no se conoce. Las dosis consumidas eran consideradas farmacológicamente relevantes (alcanzando 500-750 mg/kg/día), estando dentro del rango de aquéllas en estudios de discriminación, pudiendo indicar, por tanto, un efecto de refuerzo central. Sin embargo, el efecto salado del GHB podría haber influido en los efectos observados. Por tanto, los resultados no son muy concluyentes.
Por otra parte, ratones que reciben GHB por vía intravenosa en respuesta a la introducción del hocico por un agujero responden con más introducciones del hocico que aquellos animales que reciben GHB de forma pasiva y que únicamente reciben vehículo como respuesta a la introducción del hocico en el agujero38.
En conjunto, los estudios de discriminación de drogas, de preferencia de plaza y de autoadministración indican que GHB podría tener un efecto de refuerzo y por tanto un efecto adictivo, si bien este efecto parece ser más débil que aquél producido por otras drogas de abuso como la cocaína o los opiáceos.
Los estudios sobre la tolerancia y dependencia son escasos, aunque existe evidencia de una cierta tolerancia a los efectos de disfunción motora en ratas y de una tolerancia cruzada con el etanol34. Además, una dosis anestésica de GHB administrada a ratas dependientes de etanol produce únicamente un ligero efecto sedante39.
Lo que en un principio parece difícil de compatibilizar son las propiedades de GHB para producir dependencia/abuso y su papel en el tratamiento de los síntomas de la retirada de alcohol y opiáceos o propiedades anti-craving40-42. Estudios in vitro en el área tegmental ventral han demostrado que a concentraciones similares a aquellas obtenidas tras el uso recreativo, el GHB actúa preferentemente sobre los receptores GABAB postsinápticos localizados en neuronas GABAérgicas, produciendo una hiperpolarización de la neurona, una disminución en la liberación de GABA y, por tanto, una desinhibición de la neurona dopaminérgica y un incremento en la liberación de dopamina43. Sin embargo, en dosis más elevadas (terapéuticas) el GHB podría actuar también sobre una población de receptores GABAB en la neurona dopaminérgica (que requieren una mayor CE50), dando lugar a una hiperpolarización de la neurona dopaminérgica y por tanto a una disminución en la liberación de dopamina. Este efecto podría explicar su aparente capacidad para reducir los síntomas de la retirada44. No obstante, los estudios acerca del tema aún son escasos y los mecanismos implicados no están del todo dilucidados.
La existencia de un síndrome de abstinencia o de retirada en animales no ha sido ampliamente descrita, si bien utilizando una escala de evaluación de intoxicación-retirada de etanol, Bania et al45 observaron un síndrome de retirada después de la administración de GHB cada 3 horas entre 3 y 6 días. Estudios en seres humanos, observaciones en unidades de urgencia, además de encuestas y testimonios recogidos, apuntan a un síndrome de abstinencia en seres humanos parecido a aquél producido por la retirada de alcohol o benzodiacepinas. Este síndrome aparece entre 1 y 6 horas después de la última dosis46, pudiendo perdurar hasta 15 días. Los síntomas iniciales son insomnio, ansiedad, agitación, temblor, náuseas y vómitos. A esto le sigue una inestabilidad del sistema nervioso autónomo caracterizada por diaforesis, hipertensión, temblores y taquicardia46. En caso de retirada después de un uso crónico o de dosis altas se han descritos casos de síntomas psicóticos, alucinaciones y delirio47. También existe un período de síndrome de abstinencia prolongado que dura entre 3 y 6 meses caracterizado por disforia, ansiedad, problemas de memoria e insomnio, y durante este período el riesgo de recaída o del desarrollo de dependencia a alcohol o benzodiacepinas es elevado48.
Agradecimientos
La elaboración de este artículo está financiada por el Instituto de Salud Carlos III, Red de Trastornos Adictivos (RD06/0001/0006).
La autora declara que no existe conflicto de intereses.
Correspondencia:
E. O'SHEA
Departamento de Farmacología. Facultad de Medicina.
Universidad Complutense.
Avda Complutense s/n.
28040 Madrid. España.
Correo electrónico: estheros@farm.ucm.es
Recibido: 21-05-2008
Aceptado para su publicación: 28-06-2008