












Vacunas COVID-19: desarrollo y práctica - COVID-19 vaccines: development and practice
Más datosThe new coronavirus is still a life-threatening menace, because of its changing nature and capacity to produce many mutations to bypass the immune system. The vaccination is the first effective weapon against COVID-19.
AimThe study's goal was to design a multi-epitope peptide vaccine (MEPV) for a mix of Omicron and Delta Coronavirus strains using immuno-chemoinformatics tools.
MethodsTo create the vaccine epitopes, seven proteins from the Omicron and Delta coronavirus strains were selected (ORF1a, ORF3a, surface protein, membrane protein, ORF7a, ORF8, and nucleocapsid protein). Antigenicity, toxicity, and allergenicity of the epitopes were evaluated.
ResultsThe designed vaccine is made up of 534 amino acids that are homogeneous, antigenic, and non-toxic. Sticky restriction enzymes (XhoI and XbaI) were used to incorporate the MEPV into the pmirGLO luciferase vector. SnapGene server was used to create primers for PCR testing. Developing the MEPV is a terrific cost-effective strategy. The created MEPV's physiochemical properties have been determined to be basic, hydrophobic, and stableImmunogenicity and immune response profiles of the developed vaccine candidate were better assessed using in silico immunological simulations.
ConclusionsWe advocate moving the built vaccine to the biological validation step, where it may test our findings using appropriate model organisms.
el nuevo coronavirus sigue siendo una amenaza mortal debido a su naturaleza cambiante y su capacidad de producir muchas mutaciones para eludir el sistema inmunitario. La vacunación es la primera arma eficaz contra el COVID-19.
Objetivoel objetivo del estudio era diseñar una vacuna peptídica multiepítopo (MEPV) para una mezcla de cepas de Omicron y Delta Coronavirus utilizando herramientas inmunoquimioinformáticas. Métodos: Para crear los epítopos de la vacuna, se seleccionaron siete proteínas de las cepas de coronavirus Omicron y Delta (ORF1a, ORF3a, proteína de superficie, proteína de membrana, ORF7a, ORF8 y proteína de nucleocápside). Se evaluaron la antigenicidad, toxicidad y alergenicidad de los epítopos.
ResultadosLa vacuna diseñada está compuesta por 534 aminoácidos que son homogéneos, antigénicos y no tóxicos. Se usaron enzimas de restricción pegajosas (XhoI y XbaI) para incorporar el MEPV en el vector de luciferasa pmirGLO. El servidor SnapGene se utilizó para crear cebadores para las pruebas de PCR. Desarrollar el MEPV es una excelente estrategia rentable. Se ha determinado que las propiedades fisicoquímicas del MEPV creado son básicas, hidrofóbicas y estables. Se utilizaron simulaciones inmunológicas in silico para evaluar mejor la inmunogenicidad y el perfil de respuesta inmunitaria de la vacuna candidata generada.
ConclusionesAbogamos por pasar la vacuna construida al paso de validación biológica, donde puede probar nuestros hallazgos utilizando organismos modelo apropiados.
COVID-19 is a pneumonia-causing member of the beta coronavirus family.1,2 Due to the lack of a single particular antiviral drug for SARS-CoV-2, the COVID-19's control measures are restricted to early diagnosis, and timely publication of epidemic information.3,4 Traditional vaccine development relies on cultivating pathogens, which Inactivating and injecting the disease-causing virus is a time-consuming technique.5,6
Researchers have been working on in silico methodologies based on bioinformatics to generate multi-epitope vaccines without the need to cultivate pathogens to speed up the vaccine creation process.7,8 Using epitopes for vaccines are made using several viral protein. They include both the virus's necessary components for triggering a cellular or humoral immune response, as well as unwanted components that might induce side effects.9 Multi-epitope vaccines have the potential to be effective in the battle against viral infections, making them good vaccine for clinical trials 10,11.
The SARS-CoV-2 genome has been sequenced, and researchers have looked into the intricacies of its proteins. SARS-CoV-2's whole genome is roughly 29.9 kb long.12 The genome's availability has paved the way for the development of a vaccine to combat this deadly illness.13 A total of 6–11 open reading frames are encoded by the SARS-CoV-2 genome (surface, envelope, membrane proteins, nucleocapsid phosphoprotein and open reading frames: 1ab, 3a, 6, 7a, 8, and 10). Researchers have recently been drawn to build durable and efficient vaccines against pathogenic illnesses as a result of the availability of advanced immunoinformatics tools, which also minimize the immunological experimental burden on model organisms. These immunoinformatics tools give a safe, accurate, and quick technique to create a multi-epitope vaccination.14,15
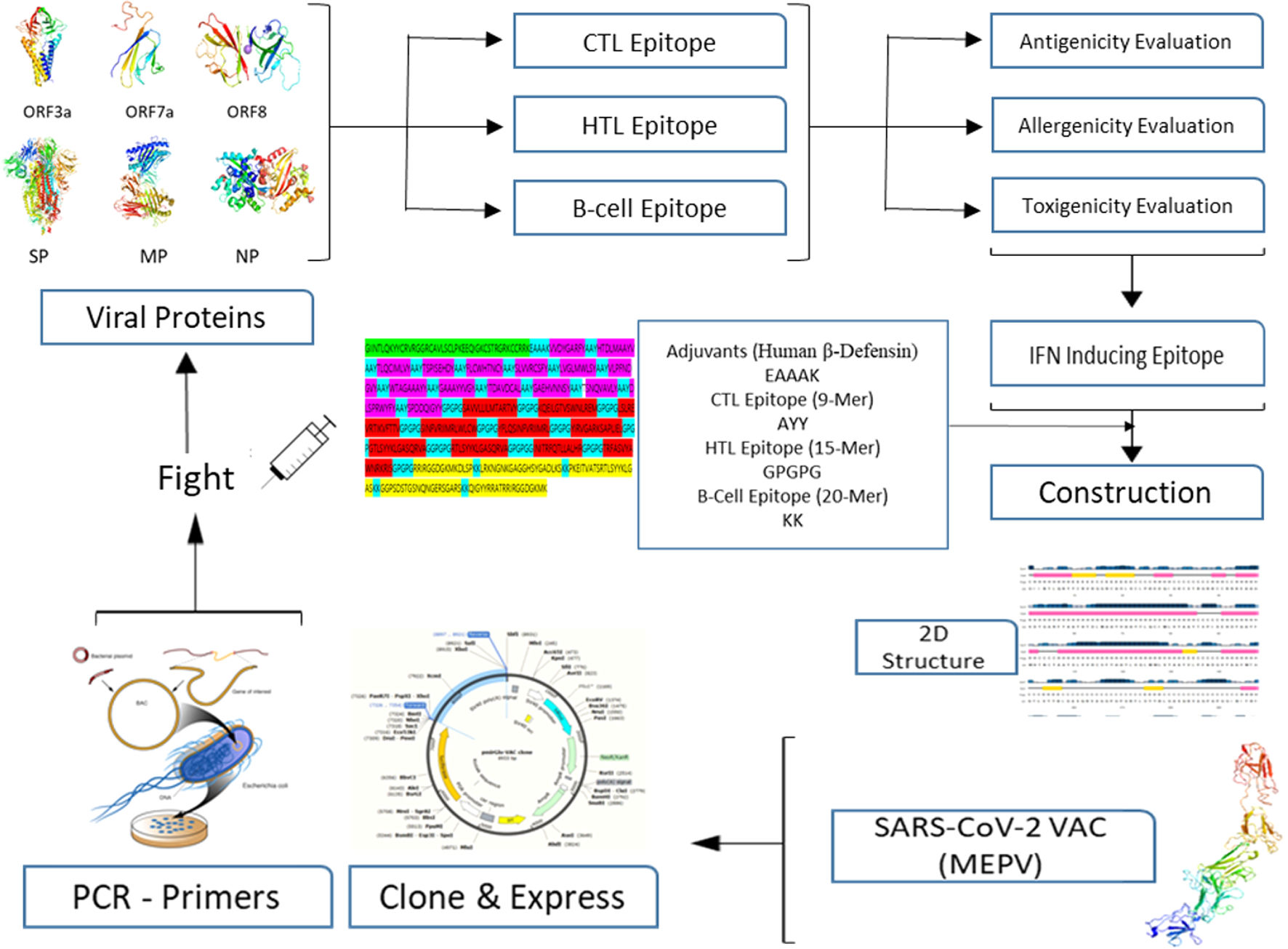
A conventional in silico vaccine design technique is shown schematically in Fig. 1. To anticipate CTL, HTL, and B- cell epitopes on entire viral proteins, scientists commonly employ a variety of in silico methods. It is also required to assess the antigenicity and other physio-chemical features of proteins. To choose the finest viral protein areas for building an effective vaccine, we must carefully and exhaustively assess all expected findings, which incurs a significant overhead and is time demanding.16,17

No present program can do many forecasts and evaluate the data completely for us all at once, allowing us to quickly pick the best vaccine components for future development and valuation. To tackle the foregoing obstacles of in silico vaccine design, we suggest using a deep neural network (DNN) architecture to make many essential predictions and extensive assessments.18,19
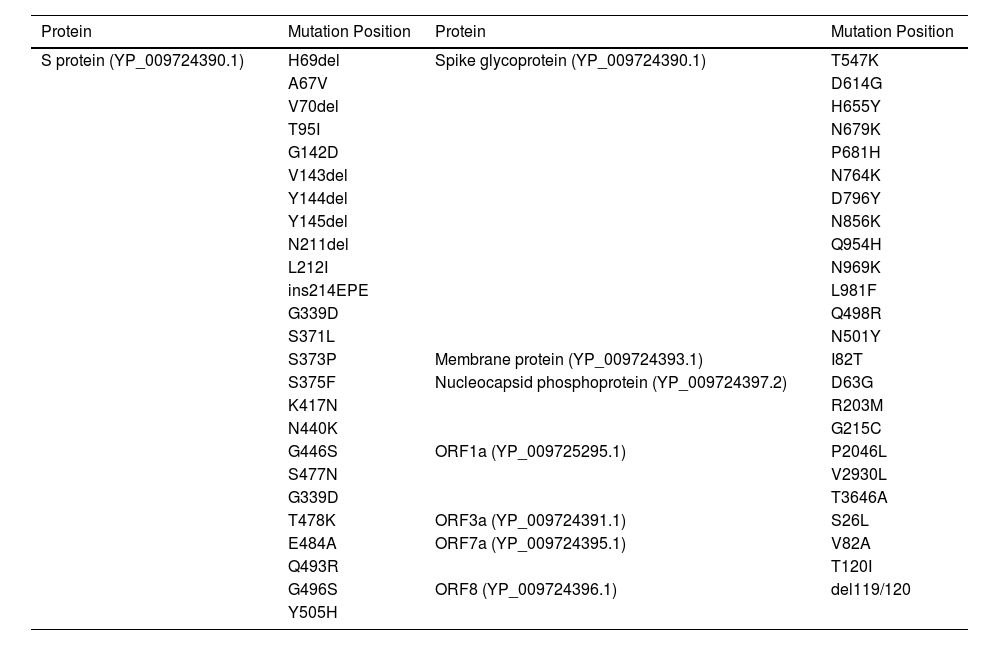
MethodsThe Omicron variant genome of SARS-CoV-2 was selected via literature from GISAID data base carrying the code (B.1.1.529). The genome of Delta SARS-CoV-2 was selected via literature from NCBI (B.1.617.2 lineage). Seven proteins were selected for the prediction of candidate vaccines: ORF1a, ORF3a, surface protein, membrane protein, ORF7a, ORF8, and nucleocapsid protein. Mutations were raised on the following proteins under study as shown in Table 1. The following diagram contributes complete designing multi-epitope peptide vaccine (MEPV) against SARS-CoV-2 Omicron and Delta variants using immuno-chemoinformatics tools.
Omicron and Delta variants of SARS-CoV-2 proteins show 48 mutation residues.
| Protein | Mutation Position | Protein | Mutation Position |
|---|---|---|---|
| S protein (YP_009724390.1) | H69del | Spike glycoprotein (YP_009724390.1) | T547K |
| A67V | D614G | ||
| V70del | H655Y | ||
| T95I | N679K | ||
| G142D | P681H | ||
| V143del | N764K | ||
| Y144del | D796Y | ||
| Y145del | N856K | ||
| N211del | Q954H | ||
| L212I | N969K | ||
| ins214EPE | L981F | ||
| G339D | Q498R | ||
| S371L | N501Y | ||
| S373P | Membrane protein (YP_009724393.1) | I82T | |
| S375F | Nucleocapsid phosphoprotein (YP_009724397.2) | D63G | |
| K417N | R203M | ||
| N440K | G215C | ||
| G446S | ORF1a (YP_009725295.1) | P2046L | |
| S477N | V2930L | ||
| G339D | T3646A | ||
| T478K | ORF3a (YP_009724391.1) | S26L | |
| E484A | ORF7a (YP_009724395.1) | V82A | |
| Q493R | T120I | ||
| G496S | ORF8 (YP_009724396.1) | del119/120 | |
| Y505H |
The antigenicity of protein sequences was assessed utilizing the (VaxiJen 2.0) server before commencing the development of the reverse vaccination candidate.
Prognosis of CTL epitopesThe prediction of cytotoxic T-lymphocyte is crucial for the manufacture of the subunit vaccine. To anticipate CTL epitopes, the main sequence was examined using the (NetCTL 1.2) server. This server recommends a 9-mer epitope sequence. The expected epitopes were categorized based on their overall score. The A1 supertype was selected in this study, despite the fact that the program only allows for CTL epitope predictions for 12 MHC class I sequences. The antigenicity of predicted CTL epitopes was assessed using the (VaxiJen 2:0) server. The antigenic CTL epitopes were determined toxicity using (ToxinPred) server. The antigenic and none toxicity epitopes were identified the immunogenicity analysis using (IEDB Class I Immunogenicity) tool. This approach has been tested on 9-mer molecules that are part of the same HLA class I molecule.
Prognosis of HTL epitopesTo predict HTL epitopes from a set of proteins, the IEDB MHCII tool was employed. The peptides were predicted using their percentile rank, SMM, and ANN values of less than 100 nM. Lower values in the percentile rank indicate that MHC II molecules have a high binding affinity. Ten HLA set molecules had epitopes predicted. The optimal epitope sequence length is 15-mer. The allergenicity was determined using the (AlgPred) software with a cut off 0.4. Antigenicity of none allergen predicted sequences were assessed using the (VaxiJen 2.0) tool. The toxigenicity of antigenic epitopes was then determined utilizing the (ToxinPred) server. Interferon-gamma (IFN-γ) stimulates natural killer NK cells and macrophages, resulting in an increased response to MHC antigen. Finally, no toxic epitopes were evaluated for IFN inducing epitopes using (IFNepitope) server. All predicted epitopes were identified of none overlapping sequence.
Prognosis of linear B-cell epitopesB-cell epitopes are necessary for inducing a humoral immune response, which stimulates B-cells and leads to antibody production. ABCpred servers were used to expose the antigens of B-cell epitope. For epitope identification with a 0.50 threshold value was used with a window length of 20 mer. Allergenicity and toxigenicity were performed for predicted epitopes using (AlgPred) and (ToxinPred) tools respectively.
Construction of MEPV candidates and assessment of their conservancyDifferent epitopes and adjuvant were linked to create the MEPV candidate. CTL epitopes were linked with the AAY linker, while HTL epitopes were linked with the GPGPG linker. B-cell epitopes, on the other hand, were linked using the KK linker. Human β-defensin 3 is the most effective adjuvant for SARS-CoV-2. DEFB3 has the potential to boost vaccination immunogenicity. This adjuvant sequence was regained in the UniProt database (ID: P81534) and the Protein Data Bank (PDB) (1kj6). Finally, the adjuvant's linker sequence is EAAAK.
Evaluation of antigenicity and allergenicity, and estimate physio-chemical properties of the constructed vaccineAntigenicity and allergenicity for the constructed vaccine was carried out using (VaxiJen 2.0) and (AlgPred) tools. Estimating of the physiochemical parameters was retrieved using (ProtParam tool) from (ExPASy) server.
Determination of 2D and 3D structure of the constructed vaccinePSIPRED, a web server, was used to anticipate the secondary structure of the proposed constructed vaccine. PSIPRED was used BLAST (Psi-Blast) to find sequences that shared a lot of similarities with the vaccine candidate. To create a position-specific scoring matrix, these sequences were chosen. The (RaptorX) webserver employs a template-free approach to anticipate the query proteome’s 2D structural attributes.
I-TASSER is a platform that predicts the 3D structure of peptide sequences. It is widely regarded as the finest server for predicting protein structure. The ModRefiner tool was used to refine the best-predicted models first, and then the GalaxyRefine server was used to improve these.
MEPV cloning and primer designAn optimization strategy was used to enhance recombinant protein expression. Most amino acids can be represented by many codons due to the degeneracy of the genetic code. The optimal CAI value is 1.0, with estimating the GC content. SnapGene server was used for predication vector-vaccine clone of conventional PCR. The optimized gene sequence for the MEPV was cloned in the plasmid vector primGLO. The AMUSER-1.0 software is used to design cloning primers.
Molecular interaction of the constructed vaccine with TLR3According to the interaction of an antigenic molecule with a specific immune receptor is acritical for measuring the immunogenicity. TLR3 (1ZIW) is a toll-like receptor used for protein-protein interaction was selected from the PDB. ClusPro 2, HDOCK, and PatchDock internet services were used for docking refinement. Pymol and BIOVIA discovery studio were used to illustrate the results. The binding pocket of the vaccination design with the receptors was evaluated using the CASTp web server. To test the protein's stability and flexibility, GROMACS was utilized to run MD simulations.
Simulation of the immune systemThe immunogenicity profile of the developed MEPV candidate was further characterized using in silico immunological simulations. For immunological simulations, the C-ImmSim server was utilized.
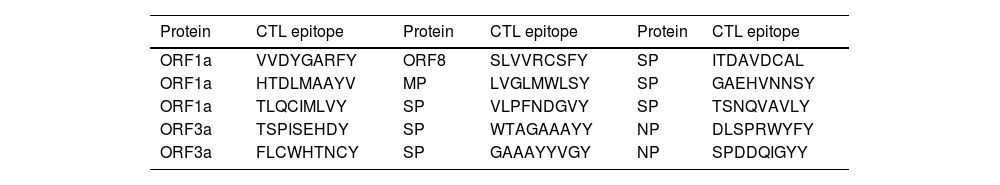
ResultsAs a result, ten CTL epitopes were selected for the final vaccine design, Table 2. Seven of them were the best sequences ranked from each strain. 2 of them were the best-conserved epitope across all strains. The last one was the best-docked epitope with HLA-A (24:02) protein.
The best antigenic, non-allergic, none toxic CTL epitopes selected from viral proteins. ORF1a: Open reading frame 1a. ORF3a: Open reading frame 3a. ORF8: Open reading frame 8. SP: Spike glycoprotein. MP: Membrane protein. NP: Nucleocapsid protein.
| Protein | CTL epitope | Protein | CTL epitope | Protein | CTL epitope |
|---|---|---|---|---|---|
| ORF1a | VVDYGARFY | ORF8 | SLVVRCSFY | SP | ITDAVDCAL |
| ORF1a | HTDLMAAYV | MP | LVGLMWLSY | SP | GAEHVNNSY |
| ORF1a | TLQCIMLVY | SP | VLPFNDGVY | SP | TSNQVAVLY |
| ORF3a | TSPISEHDY | SP | WTAGAAAYY | NP | DLSPRWYFY |
| ORF3a | FLCWHTNCY | SP | GAAAYYVGY | NP | SPDDQIGYY |
Predicted CTL epitopes were (9-mers). On the basis of, the binder locations were chosen. Then they were analyzed for antigenicity, allergenicity, Table 2. All CTL epitope peptides are linked with AAY peptides.
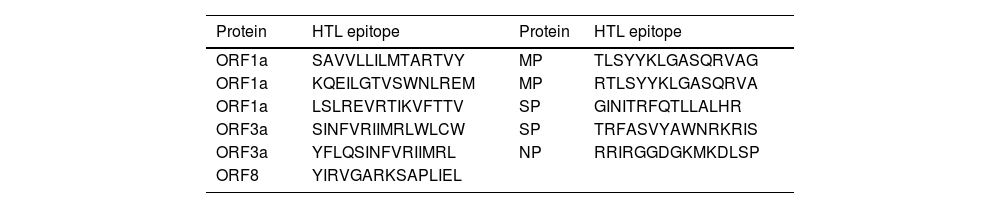
Predication of HTL epitopesMHC-II Binding Predictions from the (IEDB Analysis Resource) website were used to identify 15-mer HTL epitopes (Table 3) according to the surface accessibility. The NetMHCII 2.2 website predicted a total of seven epitopes for human alleles HLA. Epitopes were assessed to determine if they were non-allergic or hazardous peptides. Except for the penultimate one, all HTL epitope peptides are linked with GPGPG peptides.
The best antigenic, none toxic HTL epitopes selected from the viral proteins.
| Protein | HTL epitope | Protein | HTL epitope |
|---|---|---|---|
| ORF1a | SAVVLLILMTARTVY | MP | TLSYYKLGASQRVAG |
| ORF1a | KQEILGTVSWNLREM | MP | RTLSYYKLGASQRVA |
| ORF1a | LSLREVRTIKVFTTV | SP | GINITRFQTLLALHR |
| ORF3a | SINFVRIIMRLWLCW | SP | TRFASVYAWNRKRIS |
| ORF3a | YFLQSINFVRIIMRL | NP | RRIRGGDGKMKDLSP |
| ORF8 | YIRVGARKSAPLIEL |
The epitope with cutoff (0.5) with the surface accessibility to pick 20-mer B-cell epitopes. B-cell epitopes were evaluated as none allergic or toxic peptides. From all viral proteins, the 4 greatest B-cell epitopes were chosen, Table 4. Except for the penultimate one, all B-cell epitope peptides are linked with KK peptides.
Conservancy of MEPV analysis15 CTL, 11 HTL, and 4 B-cell epitopes were included in the final MEPV design. Fig. 1 shows how AAY, GPGPG, and KK peptides were used to link the anticipated epitopes. The human β-defensin adjuvant was utilized as the initial portion of the vaccine to boost immune response. This adjuvant is made up of 44aa used against HIV-1. Adjuvants with CTL epitopes are linked using the EAAAK sequence, Fig. 2.
Determination of physio-chemical and solubility properties. Adjuvants. CTL epitopes. HTL epitopes. B-cell epitopes. Linkers.")





The (ProtParam tool) was used to extract physiochemical parameters from the (ExPASy) service. The molecular weight of the finished protein was calculated to be 31.9 kDa, with isoelectric point score of 4.28. In vitro, the half-life was estimated as 100 hours in mammalian reticulocytes, and around 10 hours in E. coli. The protein's instability was 38.01, indicating that it is exceptionally stable. The thermos ability 65 is indicated by the high aliphatic index score of 77.11. The hydropathicity Grand Average was discovered (-0.046). Negatively charged residues (aspartate and glutamate) account were 23 of the total, whereas positively charged residues (arginine and lysine) account were 49.
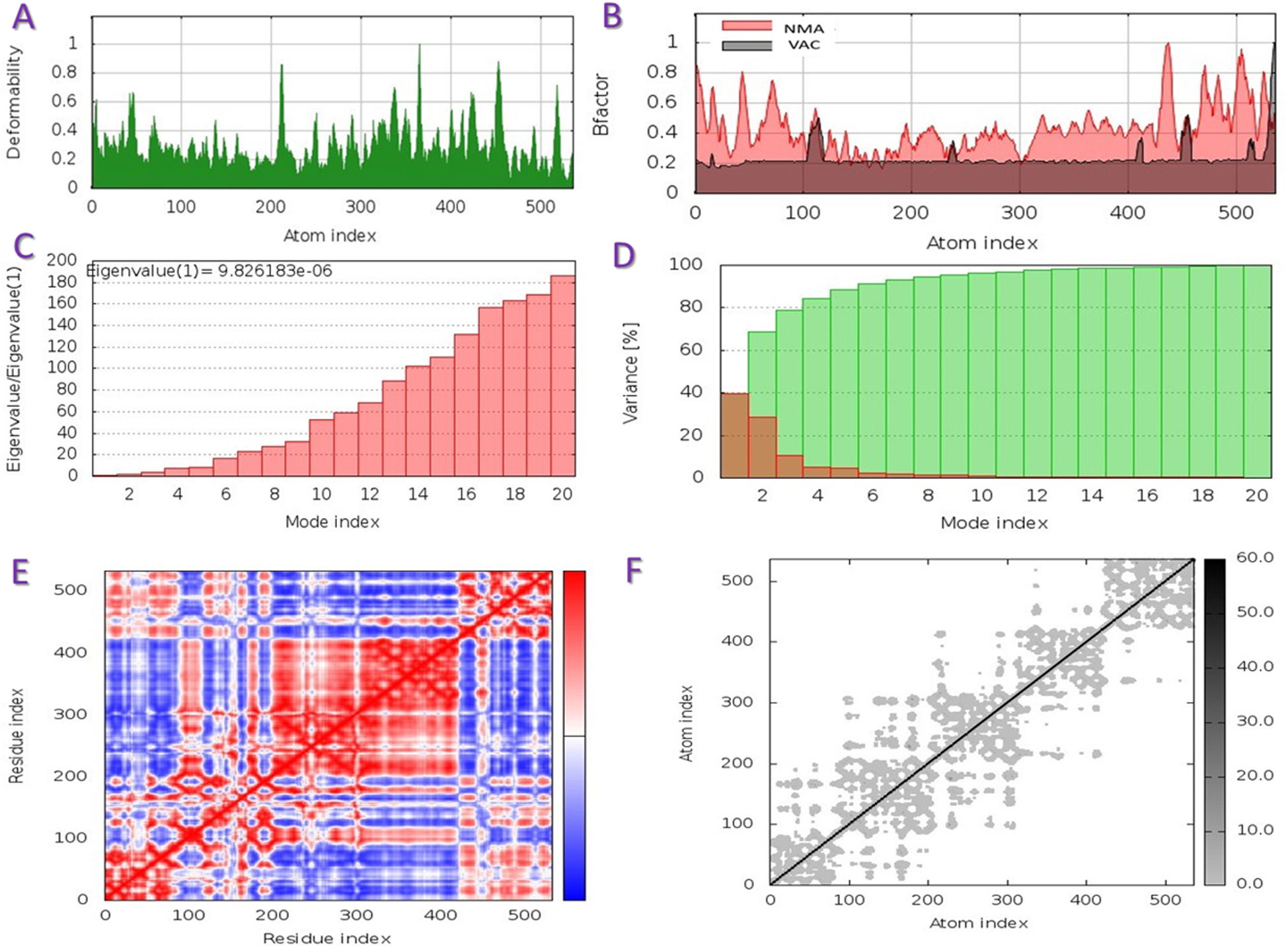
The capacity of a particular molecule to deform the residues is measured by its main-chain deformability. High deformability zones may be used to fig. out where the chain 'hinges' should be as shown in Fig. 3-A, where the majority of the residues have deformability of less than 0.3. The experimental B-factor is retrieved from the sequence field that corresponds to it. By increasing the NMA mobility by (8pi^2), the calculation from NMA is achieved. For all anticipated residues, the B-factor column provides an averaged RMS under the NMA, Fig. 3-B. Its value is related to the estimation of energy required to distort the structure. According to the observation, the energy is high, implying that the percentage of deformation is relatively low, Fig. 3-C. The eigenvalue is inversely proportional to the variance of each normal mode. Colored bars represent both individual (brown) and cumulative (green) variations, Fig. 3-D. The covariance matrix shows whether two residues are linked (red), anti-correlated (blue), or uncorrelated (white), in their movements. The majority of residues are substantially correlated, Fig. 3-E. Each dot in the fig. indicates a spring between atoms in the respective pair. The color of the dots indicates the stiffness of the springs; darker grays imply stiffer springs and vice versa, Fig. 3-F.
 and NMA. C: Energy value to deform the structure. D: Eigenvalue proportional of colored bars represent both individual (brown) and cumulative (green) variations. E: Covariance matrix shows two linked residues (red), or anti-correlated (blue), or uncorrelated (white). F: Elastic network model defines connected pairs of atoms. The darker grays indicate stiffer springs.")
Schematics physiochemical properties of the predicted vaccine. A: Deformability of residues. B: representation B-factor of peptide (VAC) and NMA. C: Energy value to deform the structure. D: Eigenvalue proportional of colored bars represent both individual (brown) and cumulative (green) variations. E: Covariance matrix shows two linked residues (red), or anti-correlated (blue), or uncorrelated (white). F: Elastic network model defines connected pairs of atoms. The darker grays indicate stiffer springs.
IFN-inducing epitopes were determined separately for the adjuvant and primary vaccination peptides because the limitation on residues numbers that may be utilized as input in the IFNepitope software. There were 51 possible IFN-inducing epitopes (25-mer) determined for the adjuvant. There were 534 possible 25-mer epitopes predicted for the primary vaccine peptide, with 73 of them scoring positively.
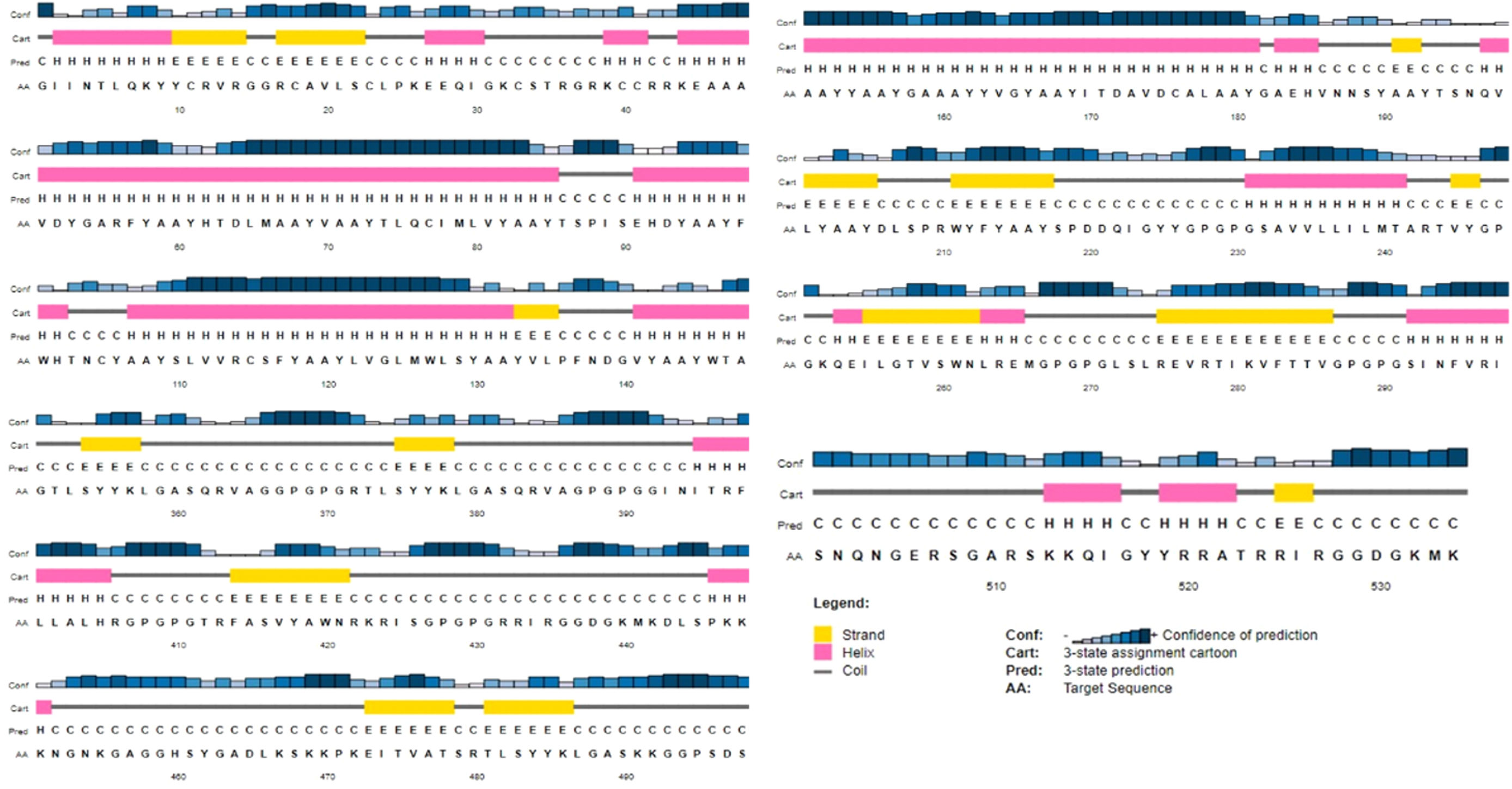
Identification of 2D & 3D structure of the constructed vaccineFig. 4 shows the 2D of the final MEVP, which was projected to feature 21% alpha-helix, 19% beta-sheet, and 59% coil. According to the PSIPRED property server, 49% of amino acid residues were exposed, 24% were medium exposed, and 26% were hidden. A total of 42 residues, or 7% of all residues, were found to be disordered. A vast set of protein physiochemical property predictors examines an incoming protein sequence, looking for things like signal peptides and other characteristics. SVM reliability was high (H) 72% when MCC for all residues in sensitivity, specificity, and precision are jointly above a given threshold, while the lowest reliability (L) was 27.3% within complete residues.
.")
The sequence scheme presented in Fig. 4 is used to map position-dependent feature predictions.
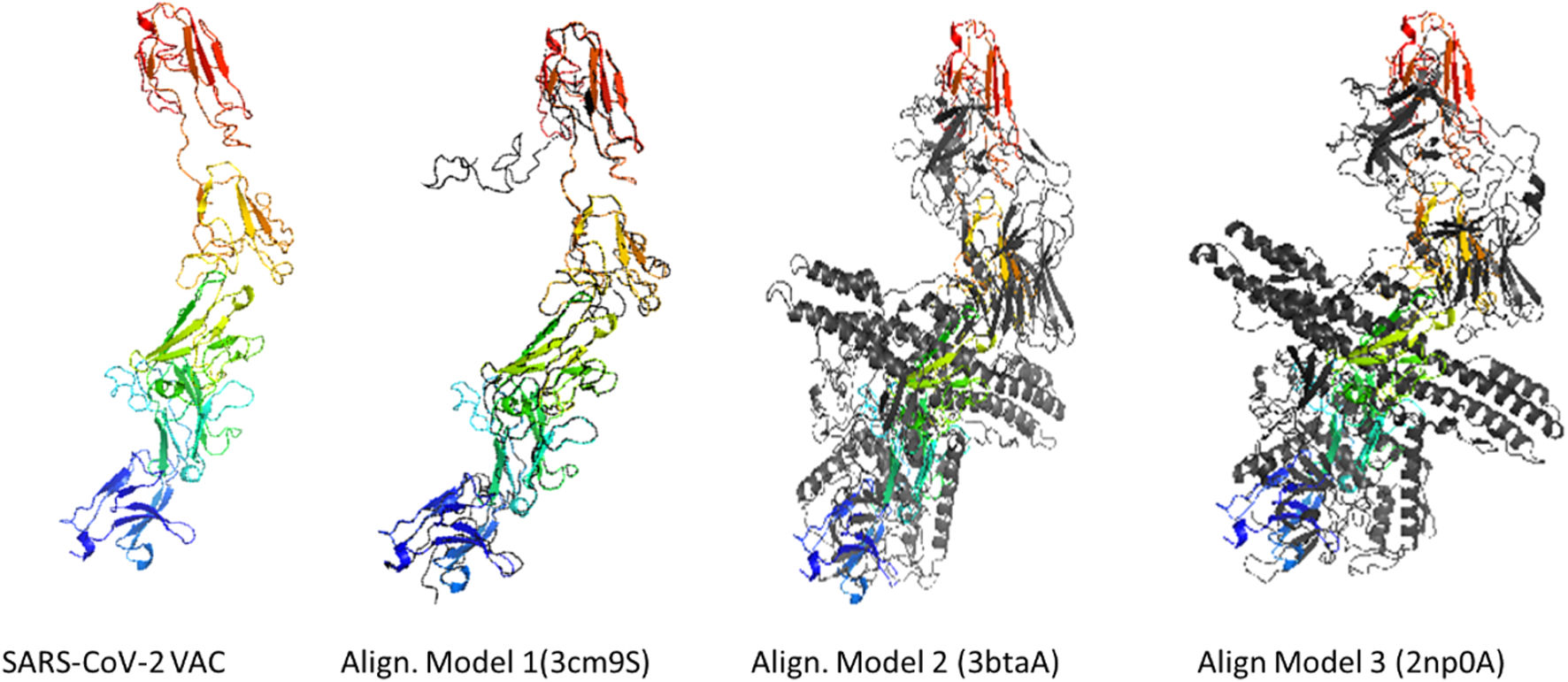
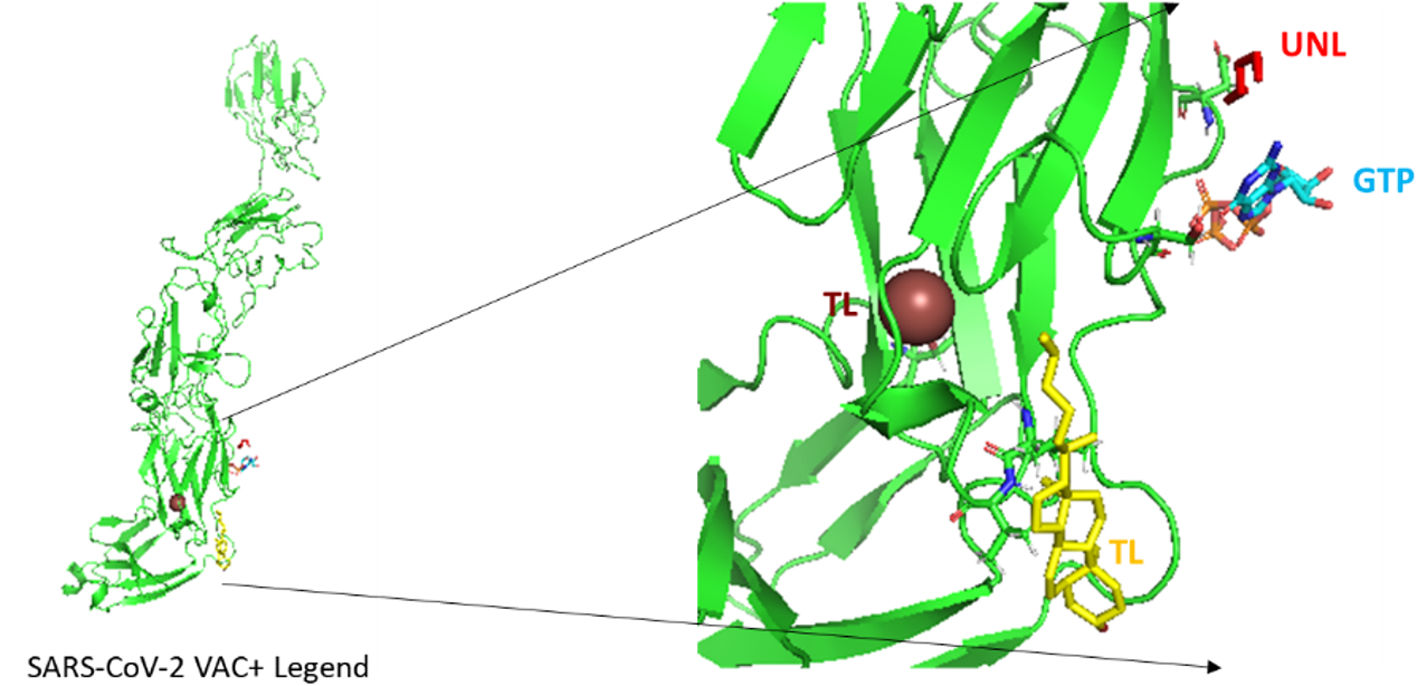
According to the I-TASSER analysis, the best predict 3D models of the vaccine candidate numerous threading alignments and reiterated structural assembly simulations. The ModRefiner tool was used to refine the best-predicted models first, and then the GalaxyRefine server was used to improve these. In the current study, we detected the highest 3 models alignment to our constructed vaccine taken from the PDB (3cm9S, 3btaA, and 2np0A). The tertiary structure of these models is observed in black cartoons aligned with the constructed vaccine (rainbow cartoon), Fig. 5. We also determined the exact residues of ligands such as guanosine triphosphate (GTP) and peptide (TL), Fig. 6.
 on the lift. Three models alignment getting from PDB (3cm9S: Human SIgA2, 3btaA: Botulinum neurotoxin serotype A and 2np0A: Mycobacterium smegmatis alpha-ketoglutarate decarboxylase homodimer) respectively.")

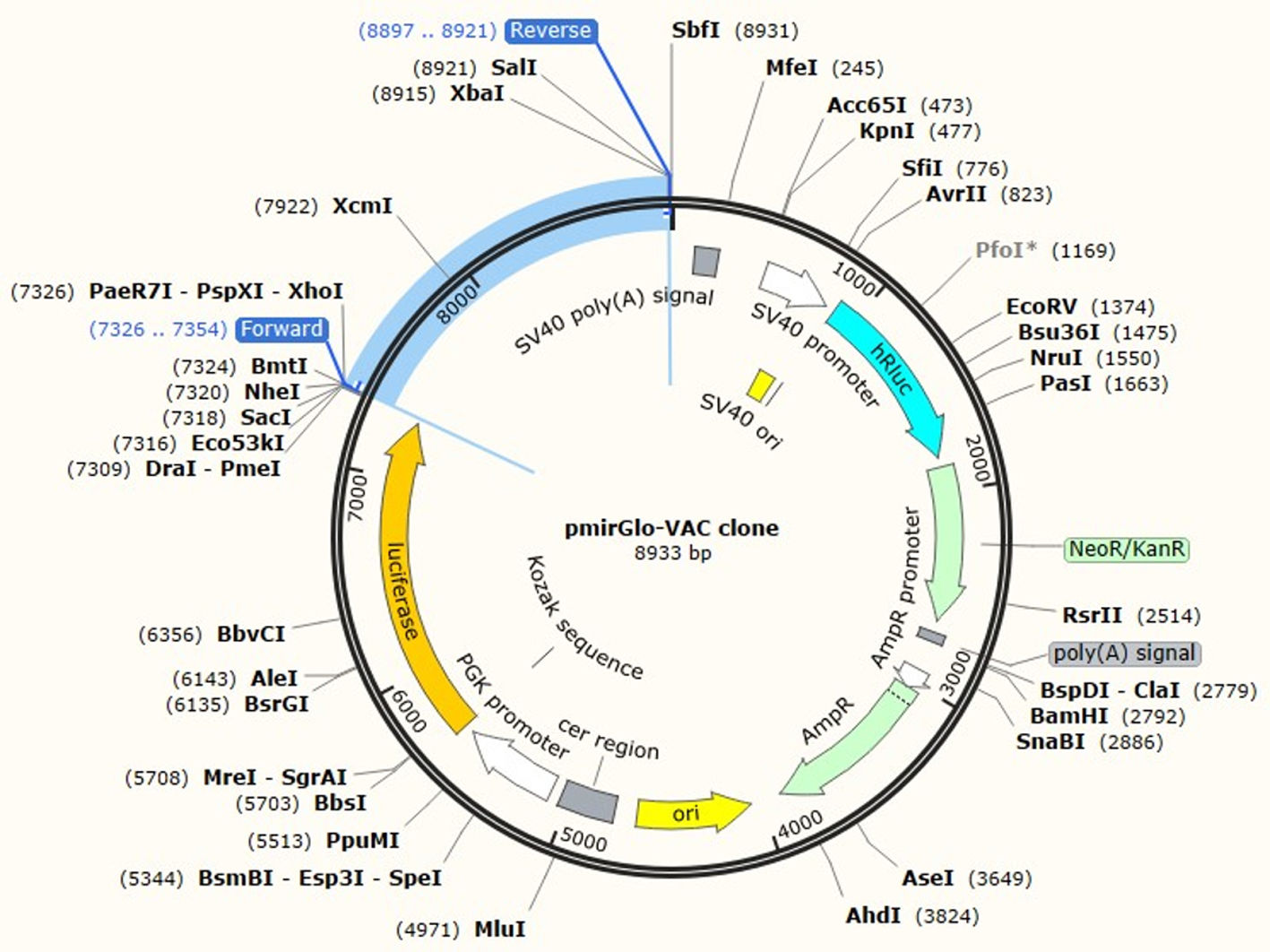
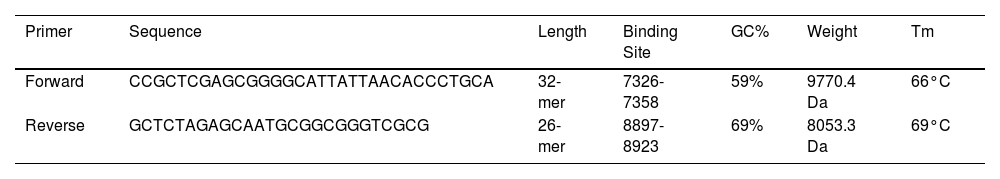
JCat software was utilized to optimize the vaccine construct's codon use for maximum protein production in E. coli (strain UTI89). The optimized sequence was 1602 nucleotides long. The average GC content of the modified sequence was 53.4%, indicating strong expression in the E. coli host. The CAI value was anticipated to be 1.0. Finally, the recombinant plasmid sequence was created by inserting the altered sequences into the plasmid vector pmirGLO luciferase using SnapGene software. XhoI and XbaI were selected to cut the vector and constructed vaccine from 5′- 3′. The appropriate forward and reverse primers were selected for conventional PCR are illustrated in the Table 5 with 59% and 69% of GC content and melting temperature between (66-69°C). Fig. 7 shows the map of constructed pmirGLO-VAC clone with 8933 bps for PCR reaction and inserted into the E. coli (strain UTI89).

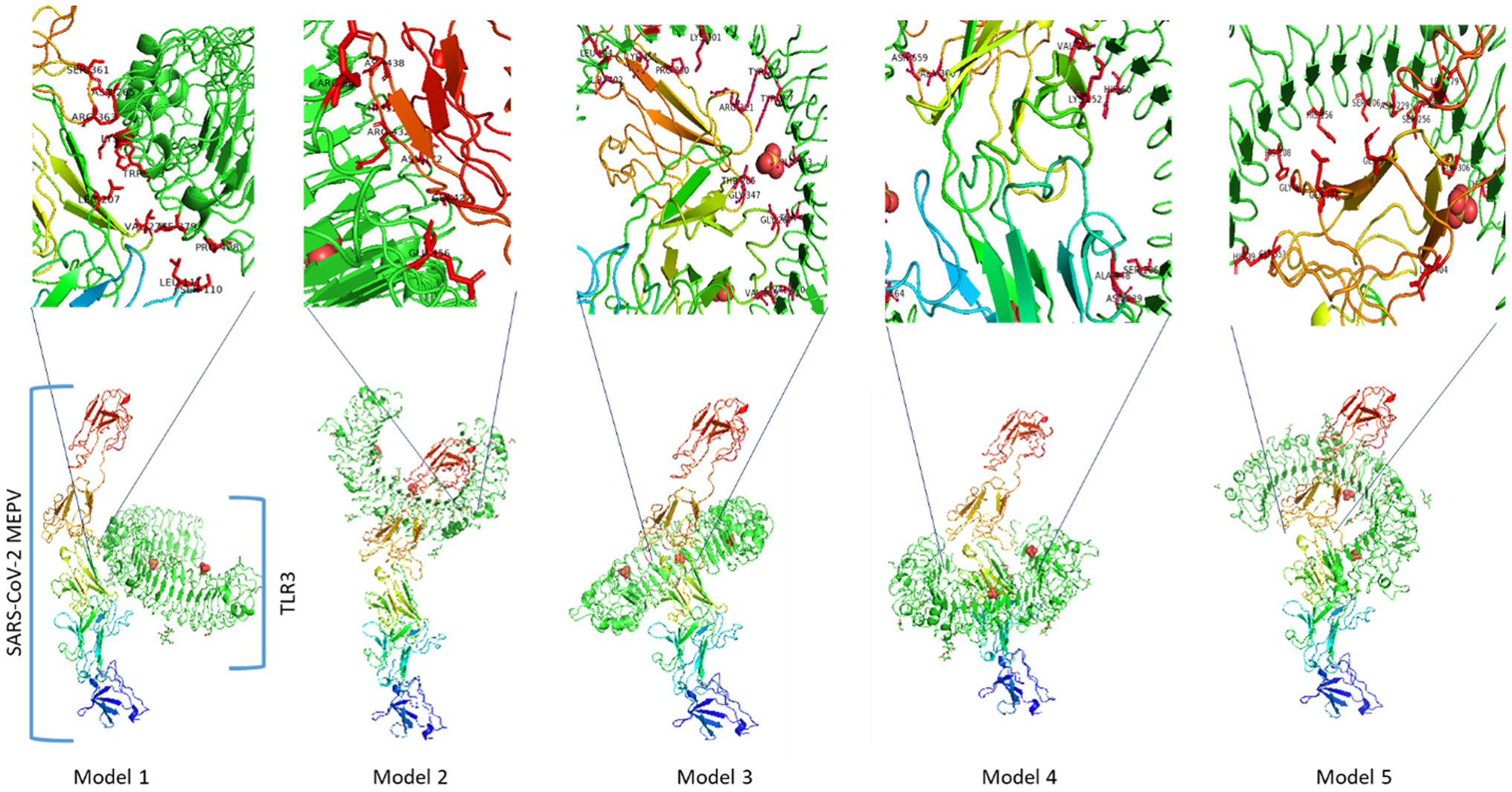
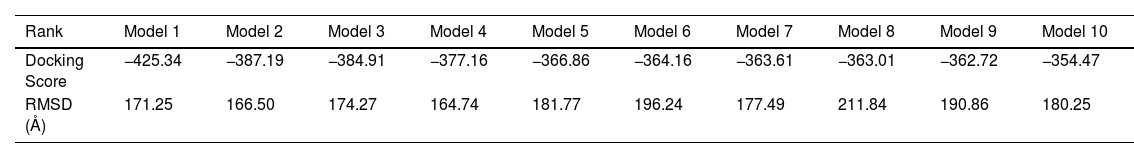
The protein interactions rise different models between the constructed vaccine and TLR3 receptor. The best 5 out of 10 docking models were drawn using different molecular docking tools, Fig. 8. According to the docking data analysis, the LGscore is very good (7.004) because it is > 5.0 while the MaxSub is correct (0.419) because it is > 0.1. There is a discrepancy in the RMSD between the predictive models, Table 6. Predicting docking models depends on the degree of interaction between proteins and the proportion of RMSD, in addition to the difference in the number of residues entering into the docking according to each predictive model.

Summary of the top 10 docking scores and RMSD models (SARS-CoV-2 MEPV-TLR3).
| Rank | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | Model 6 | Model 7 | Model 8 | Model 9 | Model 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Docking Score | −425.34 | −387.19 | −384.91 | −377.16 | −366.86 | −364.16 | −363.61 | −363.01 | −362.72 | −354.47 |
| RMSD (Å) | 171.25 | 166.50 | 174.27 | 164.74 | 181.77 | 196.24 | 177.49 | 211.84 | 190.86 | 180.25 |
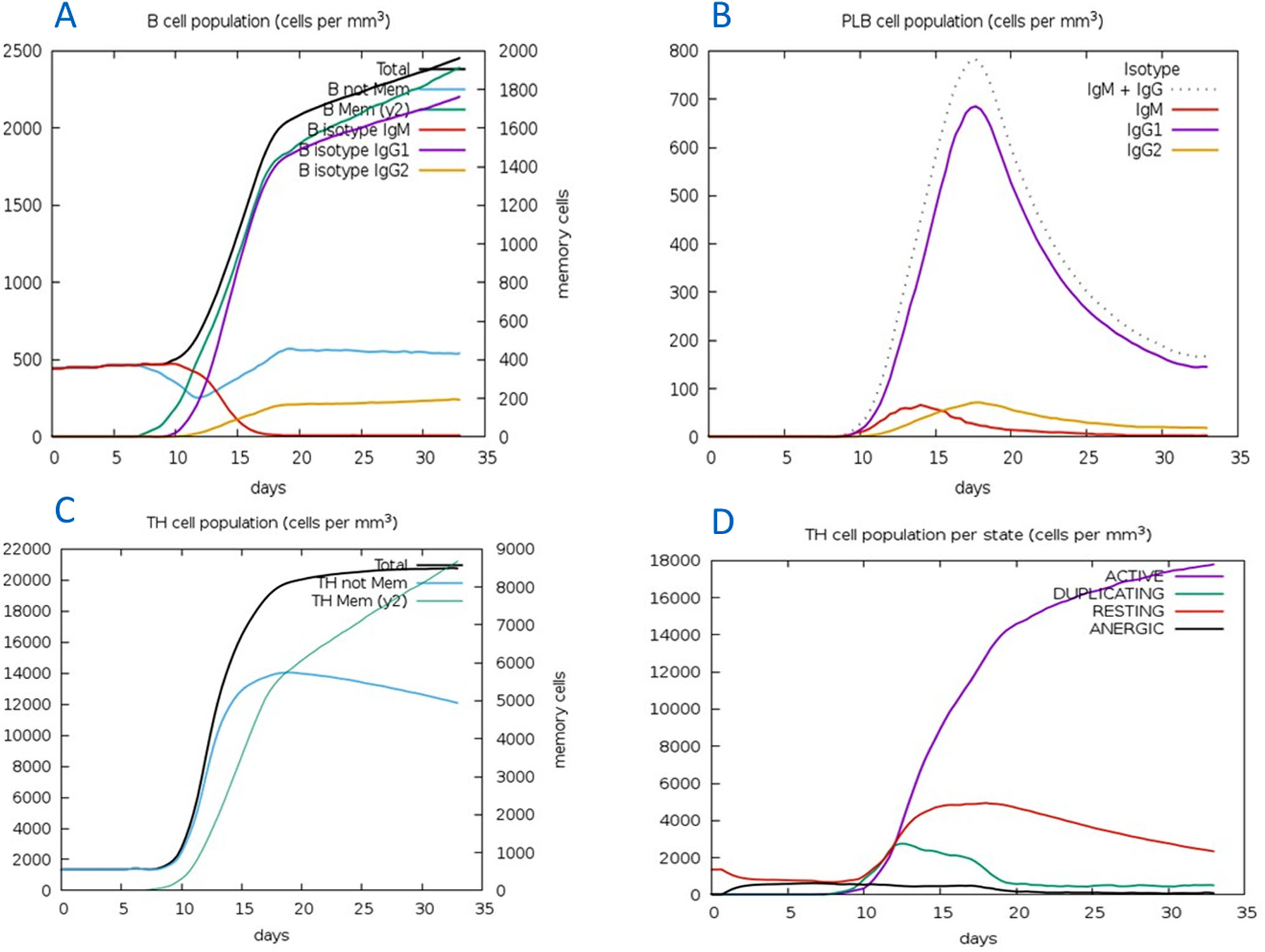
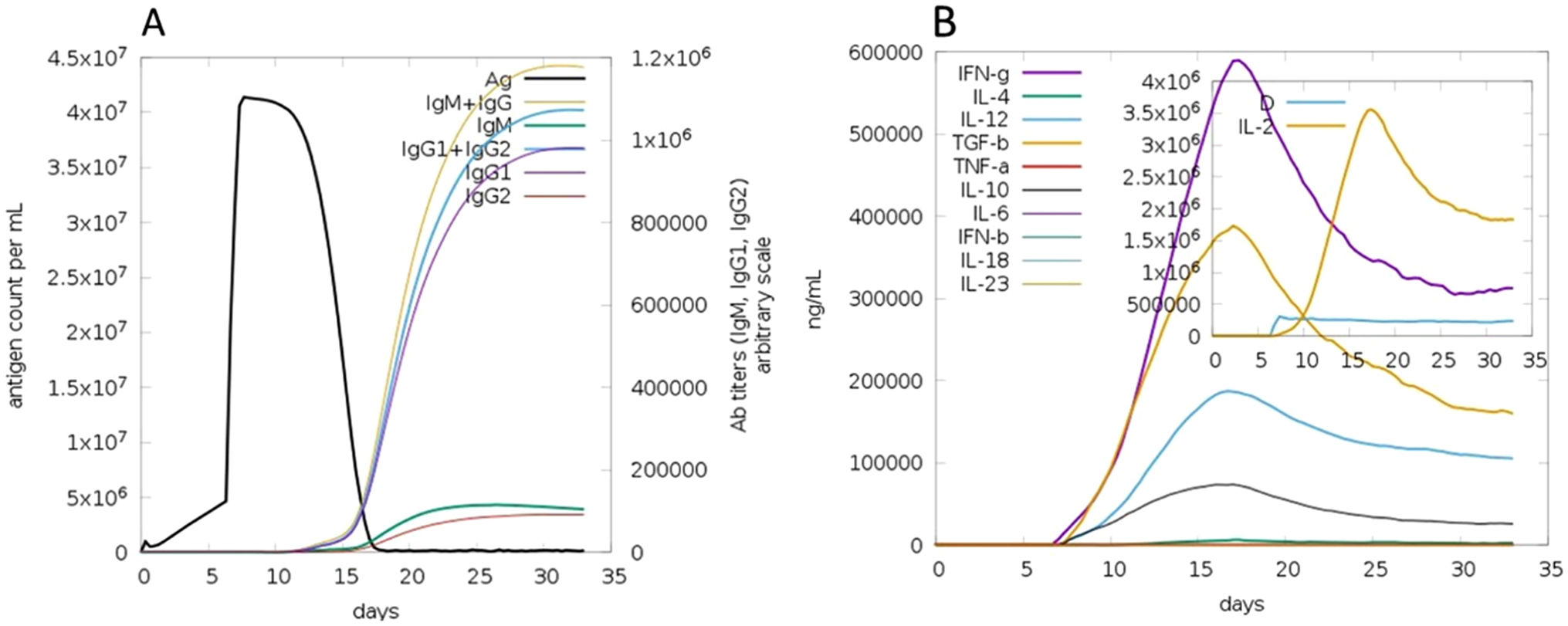
The C-ImmSim server for immune simulation was compatible with genuine immunological responses. The levels of IgG1, IgG1+ IgG2, IgM, and IgG+ IgM antibodies increased dramatically after the second and third vaccination doses. The total number of lymphocytes, antibodies, and cytokine concentrations were all graphically shown in the simulation results. The results of the simulation reveal that secondary and tertiary immune responses are more common than initial immune responses. B memory cells and IgM are primarily responsible for the initial response, as seen in Fig. 9-A, B. The concentration of T helper and TH not memory cells are considerably high after administration of the constructed vaccine, (Fig. 9-C). Activation of TH cell rises after the 10th day from the vaccine injection, Fig. (9-D). IgM+IgG and IgM titers are elevated starting from the 2nd week after taking the first dose, Fig. 10-A. Cytokines and interleukins were considerably increased. The highest level was shown for the IFN-gamma, Fig. (10-B).

In silico immune simulation with the vaccine sequence. A: B- memory cells and IgM are raised initially. B: IgM+IgG and IgG1 elevate from the 10th day after the first dose. C: T helper and TH not memory are increased in population. D: Activation of TH cell rises after the 10th day from the vaccine injection.
 VS antibody titers in different colors. B: Concentration of cytokines and interleukins in different levels. D is a diversity metric. D increases throughout time, indicating the formation of different epitope-specific dominant T-cell clones.")
The new coronavirus is still a life-threatening menace, because of its changing nature and capacity to produce many mutations to bypass the immune system.21,22 The vaccination is the first effective weapon against SARS-CoV-2. The traditional techniques to vaccine development have certain issues with vaccination efficiency and expression. To conquer all challenges, emerging techniques such as "epitope-based vaccination" appear to be more promising than full protein vaccines.23 The availability of computational tools makes it simple to create subunit-based vaccines that are antigenic and capable of eliciting an immune response. Immuno-chemoinformatics is crucial in the development of safe, stable, efficacious vaccines for a human immune response.24
In the current study, we designed the vaccine from different viral proteins (ORF1a, ORF3a, surface protein, membrane protein, ORF7a, ORF8, and nucleocapsid protein). The formation of multi-epitope vaccine (MEPV) requires the detection of CTL, T-helper cell HTL, and B-cell epitopes. Multiple sequence analysis was used to identify conserved epitopes, followed by predictions of HTL, B-cell, and IFN -γ epitopes. There were no overlapping sequences detected when the operation was completed. Antigenicity, allergenicity, and toxicity tests were performed on these epitopes. By stimulating T-helper cells to be exceptionally successful in generating robust immunoreactions, the vaccine design can improve the IFN-reaction. Epitope-based vaccines may benefit from conserved epitopes and host-specific non-homological epitopes. Around 30% of the predicted epitopes in our analysis are found in defined regions. We revealed that the percentage of predicted epitopes is very low at the site of mutations because these epitopes were toxic or allergy stimulating. To improve immunogenicity, an adjuvant human-defensin was used in the creation of this multi-epitope vaccine, which was coupled to the EAAAK linker in the N-terminal location.
This work leads to the development of a new multi-epitope vaccination against the Omicron and Delta SARS-CoV-2 variant with a length of 534aa. It includes an adjuvant as well as 15 CTL epitopes, 10 HTL epitopes, and 5 B-cell epitopes in subunits. It has strong antigenicity, population coverage, structures, and physiochemical characteristics. It has a lot of promise for the next stage of COVID-19 vaccine creation, which will involve actual tests and clinical trials.
The physiochemical features of the constructed MEPV have been determined to be basic, hydrophobic, and stable. The measured aliphatic and instability index values indicated that the vaccination protein was capable and thermostable. The positive GRAVY score designates that the vaccine site is hydrophobic. This indicates that the epitopic vaccination is capable of eliciting a high immune response without causing allergic responses.25,26
The determination of 3D structure provides a thorough understanding of how key protein components are arranged spatially. It's ideal for studying protein activities, protein components, ligand interactions, and dynamics.27 The MEPV structure's desired characteristics have improved significantly after modification.
The finished structure was then docked to TLR3 to see if it has ability binding to the immediate immunological response. TLR3 is thought to improve the antiviral process of infection and aid in the detection of dead infectious cells. The significant binding affinity of SARS-CoV-2VAC-TLR3 is required to deliver the vaccine into the body efficiently. To ensure optimal structural stability, minimizing energy was carried out to reduce the amplitude energy of the docked VAC-TLR3 complex. The eigenvalue obtained demonstrates the stiffness of the movement as well as the energy required for complicated deformations.28,29
In addition, the JCA tool was utilised to optimise the vaccine's mRNA in order to ensure the planned vaccine's translation efficiency in a particular expression system.30 XhoI and XbaI restriction enzyme cutting sites were subsequently added to the N and C terminals, respectively. After that, the vaccine sequence was cloned into the pmirGLO luciferase expression vector.
Based on the immunological results, we may conclude that the proposed vaccine can boost antibody production 10 days after the first dose is administered.31,32 Furthermore, the synthesis of interferon, as well as various interleukins and cytokines, stimulates the immunological response. Also, considerably activating immune cells has a considerable effect in limiting viral transmission, as evidenced by the proportion of more than 300 amino acids of the vaccine that could cross the host cell's transmembrane. When carrying out the vaccine laboratory, the cohesiveness of the vaccine subunits in a high percentage would be beneficial.33,34
Immunoinformatics techniques were used by Baruah and Bose to uncover CTL and B-cell epitopes for the SARS-CoV-2 spike protein. Abraham et al. created a MEPV that stimulates both CD4+ and CD8+ of T-cell immunological responses.35,36 He was built up the peptide using immunoinformatics tools. The majority of these studies are limited to vaccines based on spike proteins.37–39
The aim this study was to set a precedent in the enhancement of MEPV vaccines against Omicron and Delta SARS-CoV-2 variants. We observed that all viral proteins must be included in the vaccine design, and the estimated vaccine size should not exceed 700 amino acids. According to this study, the engineered vaccinations might be evaluated in vivo and in vitro to produce a potential SARS-CoV-2 infection vaccine for all variants. The suggested vaccine design methodology may be used to address the most often identified mutations as well as additional possibly undiscovered variants.
ConclusionsBefore biological studies, relying on contemporary in silico methodologies for vaccine development is a critical methodology that may direct research with a high possibility of producing an effective vaccine with fewer trials. The development of a multi-epitope vaccination (MEPV) is a fantastic cost-effective approach. The proteome of 7 proteins of the Omicron and Delta SARS-CoV-2 variant was used in the current investigation to build a MEPV vaccine. These proteins were subjected to immuno-chemoinformatics and structural vaccinology in order to develop a MEPV that was conducted to be effective against COVID-19 using online methods. Despite the effectiveness of computer epitope prediction and confirmation, this topic requires further experimental work. We advocate moving the built vaccine to the biological validation step, where it may test our findings using appropriate model organisms.
FundingThis work was done with self-supporting.
Conflict of interestThere is no conflict of interest in this work.
Author contributionsDesigning the idea, choosing programs, writing the manuscript, using software, writing the results, analyze and clarifying the purpose of the study were done by the corresponding author.
The author thanks college of Medicine, University of Mosul for documenting this work.