






La hipercolesterolemia familiar (HF) es una de las enfermedades genéticas más frecuentes y de mayor gravedad, que invalida y acorta la vida a los pacientes que la padecen. El tratamiento hipolipemiante mejora de forma sustancial el pronóstico de los pacientes con HF y, por ello, es imprescindible que reciban el tratamiento adecuado. La Sociedad Española de Arteriosclerosis (SEA) ha sido, y es, pionera en el diagnóstico y el tratamiento de la HF y, desde sus inicios, este grupo de pacientes ha sido objeto de su máximo interés clínico y científico, y muy especialmente para las Unidades de Lípidos de la SEA, donde se atiende a la mayor parte de sujetos con HF en España. Este documento surge de la voluntad de nuestra sociedad de actualizar el conocimiento científico para ofrecer a los médicos que controlan a estos pacientes unas directrices claras con respecto al diagnóstico y al tratamiento. Estas directrices destacan 2aspectos principales: diagnosticar la enfermedad lo más precozmente posible y reducir de forma más temprana el colesterolLDL hasta valores normales. En los próximos años hay que conseguir, entre todos los agentes implicados, que la inmensa mayoría de los pacientes con HF conozcan su diagnóstico y tengan a su disposición el tratamiento adecuado.
Familial hypercholesterolemia (FH) is one of the most common and severe genetic diseases, causing disabilities and premature death to those who suffer it. Lipid-lowering therapy substantially improves the prognosis of FH patients and, therefore, appropriate pharmacological treatment is of the utmost importance. The Spanish Society of Arteriosclerosis (SEA) has always been a pioneer in the diagnosis and treatment of FH. Since its inception, FH has been one of the main areas of clinical and scientific interest, mainly for Lipids Units of the SEA, where most patients with this pathology are referred in Spain. This document arises from the willingness of our society to update the scientific knowledge on this subject and to provide physicians with clear clinical guidelines regarding diagnosis and treatment of FH. These guidelines can be summarized in two main aspects: early diagnosis of the disease and a rapid normalization of LDLcholesterol. In the coming years, health providers should accomplish that the majority of patients with FH are aware of their diagnosis and that adequate treatment is provided.
La hipercolesterolemia familiar (HF; OMIM #143890) es un trastorno genético autosómico codominante del metabolismo de las lipoproteínas caracterizado por concentraciones plasmáticas muy altas de colesterol unido a las lipoproteínas de baja densidad (cLDL), xantomas tendinosos y un riesgo muy alto de enfermedad coronaria prematura1. La penetrancia de la HF es próxima al 100%, lo que significa que la mitad de la descendencia de una persona afectada tendrá el cLDL elevado desde el nacimiento, sin diferencias entre sexos.
La causa de la HF fue descrita por los investigadores Goldstein y Brown2, quienes encontraron que la enfermedad era consecuencia de un defecto en la captación celular de las LDL. Ellos describieron el metabolismo de las partículas LDL, el receptor celular responsable de su captación (LDLr) y el gen encargado de la síntesis del receptor LDL (LDLR). Además, detallaron las primeras mutaciones en el LDLR responsables de la enfermedad. La gran mayoría de los casos de HF son causados por mutaciones en el LDLR, pero también se produce por mutaciones en la apolipoproteína (apo) B, que es el ligando en las LDL para el LDLr, o en el gen PCSK9, una serin-proteasa que regula el reciclado intracelular del LDLr. La frecuencia de mutaciones en APOB y PCSK9 causantes de HF no supera el 10% de los casos de HF en la mayoría de las poblaciones estudiadas. Además, en torno al 20-40% de los sujetos con características clínicas de HF no presentan mutaciones en estos 3genes, por lo que otros loci tienen que estar implicados en la HF o bien pueden representar formas poligénicas. Muy recientemente se ha propuesto como causa adicional de HF una mutación en el gen de la apoE (p.Leu167del), que es una deleción del aminoácido 149 de apoE3.
PrevalenciaLa HF es uno de los trastornos hereditarios más frecuentes. Se estima una prevalencia en la población en torno a 1:500 y 1:1.000.000 para heterocigotos y homocigotos, respectivamente. En algunas poblaciones con cierto aislamiento genético y elevada consanguinidad, como canadienses franceses, libaneses cristianos, sudafricanos Afrikaner, o judíos Ashkenazi, la prevalencia puede llegar a 1:100sujetos, y entonces un pequeño número de mutaciones en el LDLR son responsables de gran parte de los casos de HF4. Sin embargo, en la mayor parte de poblaciones, incluida la española, con mayor variabilidad genética, la HF es causada por cientos de mutaciones diferentes. Solamente en España se han descrito más de 250mutaciones diferentes en el LDLR como causantes de la enfermedad5.
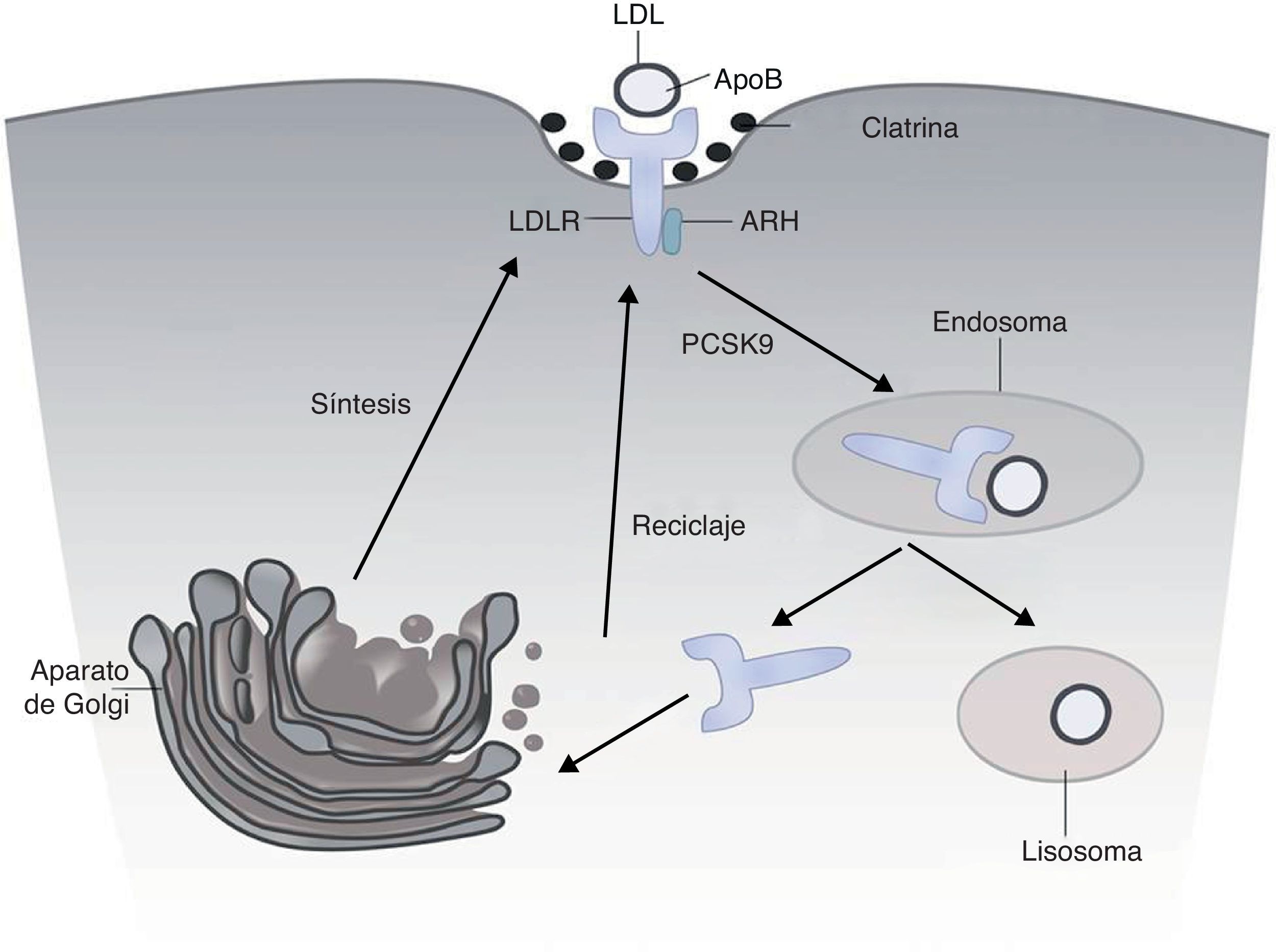
El receptor LDL (LDLr)El LDLr se sintetiza como una proteína de 120kDa y se concentra en la membrana celular en invaginaciones de la membrana recubiertas de clatrina. El LDLr reconoce y une a las apoB de las LDL y también a la apoE de las partículas remanentes. Tras un complejo proceso de endocitosis migra a los endosomas, donde por la acidificación de los endosomas la partícula LDL se metaboliza, y el LDLr se recicla a la superficie celular (fig. 1).
 se sintetiza como un precursor de 120kDa y se procesa en el aparato de Golgi, desde donde es transportado a la superficie celular a las invaginaciones recubiertas de clatrina de la membrana. Allí se une a la partícula LDL. El complejo se transporta a los endosomas, donde se disocia y se libera el LDLr a la superficie celular. La unión de PCSK9 con el LDLr acelera su degradación lisosomal.")
El receptor LDL (LDLr) se sintetiza como un precursor de 120kDa y se procesa en el aparato de Golgi, desde donde es transportado a la superficie celular a las invaginaciones recubiertas de clatrina de la membrana. Allí se une a la partícula LDL. El complejo se transporta a los endosomas, donde se disocia y se libera el LDLr a la superficie celular. La unión de PCSK9 con el LDLr acelera su degradación lisosomal.
La producción del LDLr está estrechamente regulada por un mecanismo de retroalimentación en respuesta a las variaciones en la concentración intracelular de esteroles y la demanda celular de colesterol. El gen LDLR modifica su expresión en respuesta a la concentración de esteroles gracias a la presencia en su zona promotora de secuencias, denominadas sterol regulatory element (SRE), que interactúan con el factor de transcripción sterol regulatory element-binding proteins (SREBP-2), un sensor intracelular de esteroles.
El gen del receptor LDL (LDLR)El gen que codifica el receptor de LDL se divide en 18exones. El exón1 codifica el péptido señal de 21aminoácidos necesario para el transporte a la superficie celular. Los exones 2 al 6 codifican la región de unión a apoB y consta de 7repeticiones en tándem de 40aminoácidos cada una. Los exones 7 al 14 codifican la zona con alta homología con el precursor factor de crecimiento epidérmico (EGF). Este dominio es necesario para la disociación de las LDL del LDLr en el endosoma y posterior reciclado del receptor. El exón15 es un dominio de glucosilación cuya función es desconocida, pero se cree que se relaciona con la estabilización del receptor. El exón16 codifica el dominio transmembrana del receptor esencial para el anclaje de la LDLr a la membrana celular, y los exones 17 y 18 codifican el dominio citosólico. El receptor de LDL puede ser descrito como una proteína quimérica. Se compone de una serie de dominios funcionalmente distintos que pueden funcionar independientemente uno del otro.
Hoy en día se han descrito más de 1.000mutaciones en LDLR en pacientes con HF a lo largo de muchas poblaciones (http://www.ucl.ac.uk/fh;http://www.umd.necker.fr). Las mutaciones causantes de HF en el LDLR se han clasificado en 5clases funcionales6.
Las mutaciones clase1 se conocen como «alelos nulos». En estos tipos de mutaciones no se produce proteína funcional. Las mutaciones clase2 son alelos defectuosos para el transporte entre el retículo endoplasmático y el aparato de Golgi; estas mutaciones se encuentran dentro de los exones que codifican la unión del ligando y el precursor EGF. Las mutaciones clase3 son alelos defectuosos en las que el LDLr se sintetiza y se transporta a la superficie celular, pero no es capaz de unirse a las partículas de LDL. Las mutaciones clase4 se conocen como alelos defectuosos para la internalización del complejo LDL-LDLr. Finalmente, las mutaciones clase5 codifican los LDLr que no son capaces de liberar las partículas de LDL en los endosomas, evitando que el LDLr se recicle a la superficie celular. Parte de la heterogeneidad en las manifestaciones clínicas de la HF se atribuye al tipo de mutación responsable. Recientemente, un estudio llevado a cabo por nuestro grupo, con 436pacientes españoles HF con mutaciones conocidas en el LDLR clasificados como alelos nulos o alelos defectuosos, ha demostrado que pacientes con mutaciones tipo alelo nulo muestran un fenotipo clínico más grave y mayor aterosclerosis carotídea que aquellos con mutaciones defectuosas, independientemente de la edad, el género y otros factores de riesgo lipídicos y no lipídicos7.
Otros genes asociados a hipercolesterolemia familiarAlgunos pacientes presentan mutaciones en APOB como responsables de la HF. En estos casos, la hipercolesterolemia se denomina apoB-100 defectuosa familiar (FDB)8. La primera y principal mutación en APOB causante de HF resulta de la sustitución del aminoácido 3500 de glutamina por arginina (R3500Q).
Datos recientes revelan que, en comparación con pacientes con HF con mutaciones LDLR, los pacientes con FDB tienen el cLDL un 20-25% más bajo, responden mejor a las estatinas y tienen un menor riesgo de enfermedad coronaria.
Mutaciones en la proproteína convertase subtilisin/kexin tipo 9 (PCSK9) se asocian a HF. Al ser el tercer responsable, a la hipercolesterolemia secundaria a PCSK9 se la conoce con HF3. PCSK9 es una proteína sérica que se une al LDLr en la superficie celular y favorece su degradación en el lisosoma. Las mutaciones en PCSK9 que producen ganancia de su función se asocian con una disminución en el número de LDLr en la superficie celular debido a un menor reciclado del mismo y un fenotipo clínico semejante a la HF9. En un estudio reciente realizado por Talmud et al.10 se ha sugerido que muchos de los pacientes en los que no se detecta la mutación a pesar de tener criterios clínicos para el diagnóstico podrían representar una forma poligénica de la enfermedad en la que varias mutaciones de genes con efectos discretos sobre la colesterolemia se transmitieran hereditariamente de forma conjunta.
Manifestaciones clínicasLos pacientes heterocigotos suelen tener concentraciones de cLDL que doblan a la media de la población desde el nacimiento. En la edad adulta suelen encontrarse entre 250-350mg/dl en los heterocigotos y por encima de 600mg/dl en los homocigotos desde el nacimiento. Este aumento de cLDL se acompaña de un aumento del número total de partículas LDL circulantes debido a un catabolismo disminuido y, por tanto, una vida media aproximadamente doble de lo normal. Las características de las partículas LDL en la HF no se diferencian de las LDL de la población normolipémica, salvo que su concentración plasmática es superior. Las concentraciones de triglicéridos suelen ser normales o solamente discretamente elevadas y suele ser un dato diferencial con otra hipercolesterolemia genética, la hiperlipidemia familiar combinada. La concentración de colesterol de las lipoproteínas de alta densidad (cHDL) es normal.
La presencia de xantomas tendinosos es frecuente en personas mayores de 40años heterocigotos y casi constantes en la primera década de la vida en los sujetos homocigotos. Su localización más característica es el tendón de Aquiles, pero también son frecuentes en codos y extensores de los dedos de las manos. La presencia de xantomas tendinosos se asocia con un mayor riesgo de enfermedad cardiovascular prematura, especialmente en mujeres, y mayor riesgo de tendinitis. En los últimos años, y gracias a la posibilidad de tratamientos eficaces para la hipercolesterolemia desde la juventud, la prevalencia de xantomas tendinosos ha descendido mucho. El arco corneal en las primeras décadas de la vida es otro depósito lipídico superficial característico de la HF que en ocasiones se utiliza como criterio diagnóstico en algunos algoritmos.
La enfermedad coronaria es el problema clínico fundamental de la HF. Sin tratamiento, y con el estilo de vida occidental actual, aproximadamente el 85% de los varones y el 50% de las mujeres sufrirían un episodio coronario antes de los 65años de edad, y es responsable del 80% de la mortalidad total. En algunas zonas del mundo, hasta el 10% de la enfermedad coronaria prematura se debe a la HF, aunque en la mayor parte de las poblaciones es responsable del 1 al 3% de la misma. Afortunadamente, la enfermedad coronaria está disminuyendo en los últimos años, y registros procedentes del Reino Unido y Holanda nos indican una reducción superior al 50% en las últimas 2décadas11.
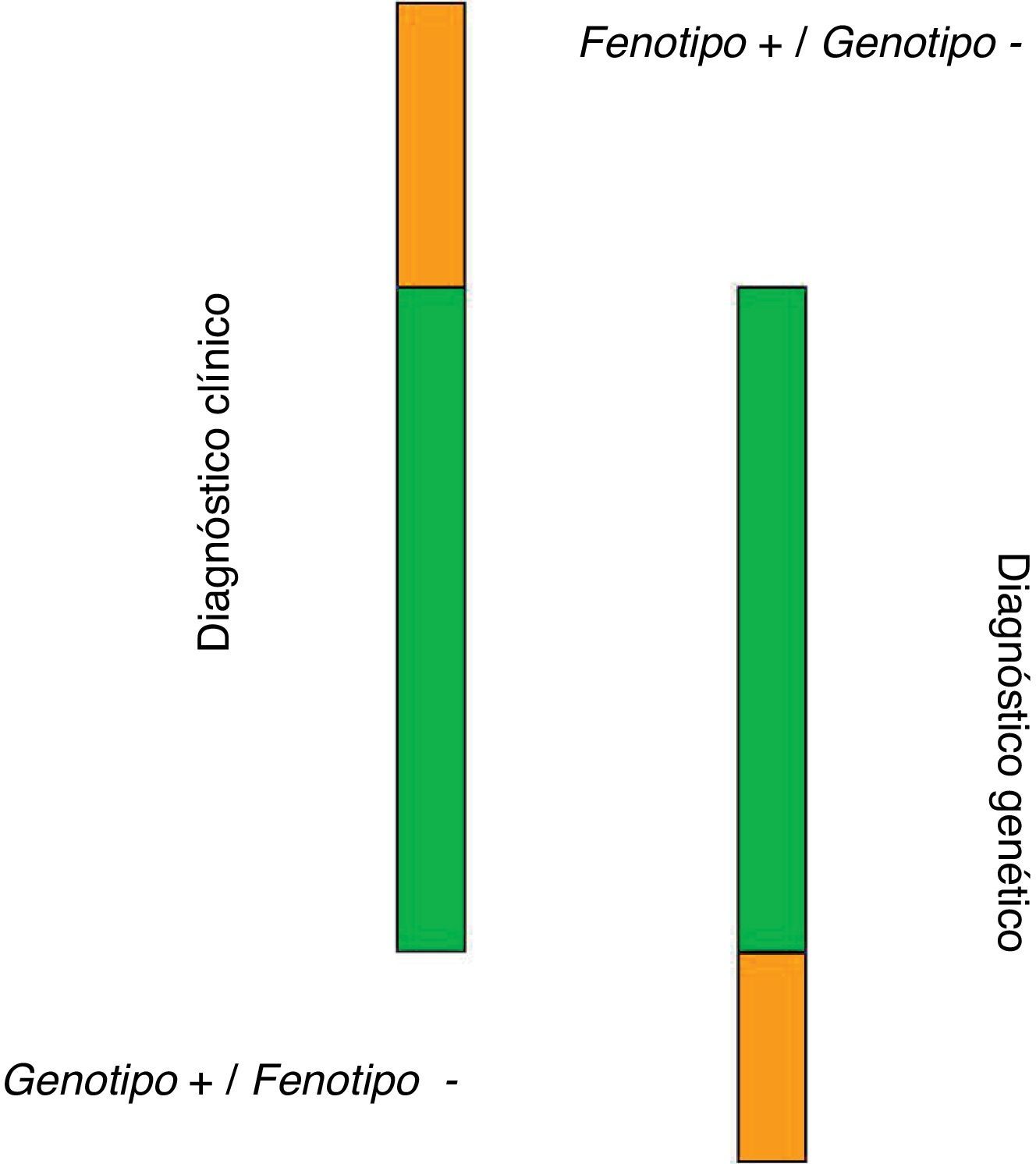
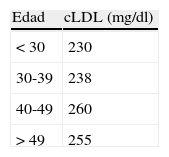
Criterios diagnósticos de la hipercolesterolemia familiarEl diagnóstico de HF se basa en criterios clínicos, bioquímicos y genéticos4. Estos son: historia personal y familiar de hipercolesterolemia severa o enfermedad cardiovascular precoz, depósitos de colesterol en forma de arco corneal o xantomas tendinosos, y presencia de una mutación funcional en alguno de los 3genes involucrados en la patogenia de la enfermedad. El diagnóstico de certeza se establece en pacientes con historia personal y familiar de hipercolesterolemia severa y la presencia de una mutación funcional. Sin embargo, esta concurrencia de datos no puede concretarse en todos los pacientes, por lo que se han establecido índices diagnósticos basados en aspectos clínicos. Los índices más utilizados son los de la red de unidades de lípidos de Holanda (DLCN) y los índices de MedPed y del registro Simon Broome. El más utilizado en nuestro medio es el DLCN, que basa el diagnóstico clínico en 5aspectos, como son la historia familiar de dislipidemia y cardiopatía isquémica, la historia personal, los datos de exploración física, los datos bioquímicos y los estudios genéticos (tabla 1). Esto permite otorgar una puntuación que estratifica la posibilidad diagnóstica en diagnóstico definitivo, probable, posible o improbable. El MedPed y el Simon Broome son de interés dado que ofrecen unos valores de corte de colesterol, total o LDL, que permiten establecer o no la sospecha clínica de HF. Civeira et al.12 definieron los puntos de corte más discriminantes para establecer la sospecha diagnóstica en distintos rangos de edad en la población española (tabla 2). Junto a la presencia de xantomas, estas concentraciones de LDL tienen un elevado grado de predictibilidad de la presencia de mutación genética. El estudio ecográfico de los tendones de Aquiles facilita la detección de xantomas tendinosos que no se perciben mediante la exploración manual, por lo que se recomienda su práctica en estos pacientes13. En España se ha desarrollado una metodología que permite el acceso relativamente rápido al diagnóstico genético basado en técnicas de secuenciación masiva que permite estudiar todas las mutaciones asociadas a la enfermedad (Liponext) (www.progenika.com) y que sustituye al antiguo microchip (lipochip). El acceso al diagnóstico genético de forma generalizada ha modificado el concepto diagnóstico de la enfermedad, dado que datos de nuestro país y otros como el Reino Unido muestran que solo en aproximadamente el 60% de los pacientes etiquetados como HF cierta (DLCN>8) se detecta una mutación responsable del trastorno5. Como se ha mencionado en el capítulo anterior, estos pacientes podrían representar una forma poligénica de la enfermedad con transmisión conjunta de varios genes10. Por otro lado, en los pacientes con escasa probabilidad de diagnóstico de HF (DLCN<3) en los que, por ejemplo, se ha realizado el test genético como parte de un cribado familiar, hasta el 16% muestran mutaciones. Ello demuestra la complejidad del trastorno al que nos referimos y sus indudables repercusiones clínicas en al menos 2ámbitos: el manejo terapéutico y la indicación de generalizar el estudio familiar. Así pues, podemos establecer 3categorías diagnósticas: a)pacientes con HF en los que los datos clínicos y genéticos son positivos; b)pacientes con datos clínicos positivos (DLCN>8) pero estudio genético negativo, y c)pacientes con mutación positiva a pesar de una clínica no sugestiva (fig. 2). El reconocimiento de esta variabilidad diagnóstica obliga a matizar la acción terapéutica y de detección del trastorno.
Criterios diagnósticos de la Dutch Lipid Clinic Network (DLCN) para la hipercolesterolemia familiar
| Puntos | |
| Historia familiar | |
| a) Familiar de primer grado con enfermedad cardiovascular precoz (<55años varón; <60años mujer) | 1 |
| b) Familiar de primer grado con cLDL>percentil 95 | 1 |
| c) Familiar de primer grado con xantomas tendinosos o arco corneal | 2 |
| d) Niños <18 años con cLDL>percentil 95 | 2 |
| Historia personal | |
| a) El paciente tiene historia de enfermedad coronaria precoz (<55años varón; <60años mujer) | 2 |
| b) El paciente tiene historia de enfermedad cerebrovascular o arterial periférica precoz (<55años varón; <60años mujer) | 1 |
| Examen físico | |
| a) Xantomas tendinosos | 6 |
| b) Arco corneal en pacientes <45años | 4 |
| Datos bioquímicos (cLDL en mmol/l [mg/dl]) | |
| >8,5 [330] | 8 |
| 6,5-8,4 [250-329] | 5 |
| 5,0-6,4 [190-249] | 3 |
| 4,0-4,9 [155-189] | 1 |
| Análisis genético ADN | |
| a) Mutación funcional en los genes LDLR, APOB o PCSK9 | 8 |
8 puntos: diagnóstico cierto; 6-7: diagnóstico probable; 3-5: diagnóstico posible; <3: diagnóstico improbable.

El diagnóstico de HF acarrea un riesgo cardiovascular alto por definición, si bien una vez diagnosticada la HF debe investigarse la presencia de otros factores de riesgo vascular adicionales, dado que su presencia colabora de forma clara en el pronóstico de los pacientes14. Obviamente, los afectados de diabetes y enfermedad cardiovascular deben ser considerados, por definición, de muy alto riesgo. Concentraciones elevadas de lipoproteína(a) [Lp(a)] se han asociado a un riesgo superior en este grupo de pacientes, por lo que su determinación es obligatoria. Se recomienda el estudio de la presencia de arteriosclerosis subclínica, determinando el índice tobillo-brazo o el grosor íntima-media carotídeo, si estas técnicas están disponibles. Así mismo, el tipo de mutación y, en concreto, las de tipo1 con presencia nula de receptor, determina un mayor riesgo cardiovascular. Estos datos facilitan la estratificación de riesgo en estos pacientes y, por tanto, la intensidad terapéutica. La tabla 3 muestra los factores que modulan el riesgo vascular en pacientes con HF.
Factores de riesgo mayores en paciente con hipercolesterolemia familiar
| Edad |
| Varón >30años |
| Mujer >45años o menopáusica |
| Tabaquismo |
| Fumadores activos |
| Historia familiar de CI precoz |
| Familiar varón 1.° con CI antes de los 55años |
| Familiar mujer 1.° con CI antes de los 60años |
| cLDL muy alto |
| Superior a 330mg/dl |
| cHDL bajo |
| Inferior a 40mg/dl |
| Hipertensión |
| Superior o igual a 140/90mm Hg o en tratamiento |
| Diabetes mellitus |
| Lp(a) alta |
| Superior a 50mg/dl |
| Mutación LDL |
| Tipo 1 alelo negativo |
CI: cardiopatía isquémica; cHDL: colesterol de las lipoproteínas de alta densidad; cLDL: colesterol de las lipoproteínas de baja densidad; Lp(a), lipoproteína(a).
Fuente: Adaptado de Civeira et al.4.
La HF es una de las alteraciones genéticas más frecuentes. Dado que desde el nacimiento las personas afectadas presentan cifras de colesterol superiores a las correspondientes para su edad y sexo, el riesgo aterógeno es muy elevado. Una detección precoz del proceso y el tratamiento temprano reducen dicho riesgo a valores similares a los de la población no afectada de HF.
La política de detección de HF puede establecerse de forma generalizada en la infancia, en la edad adulta, por cascada diagnóstica a raíz de la detección de un caso índice o de forma oportunista. En el apartado de recomendaciones nos referimos a cada una de ellas, pero de forma general. Debemos plantear el diagnóstico en las siguientes situaciones:
- 1.
Familiares de primer grado de pacientes diagnosticados de HF.
- 2.
Pacientes con historia familiar de cardiopatía isquémica precoz.
- 3.
Pacientes con historia personal de cardiopatía isquémica precoz.
- 4.
Cifras de cLDL>190mg/dl, o >160mg/dl en niños y adolescentes.
- 5.
Detección de arco corneal antes de los 45años.
- 6.
Presencia de xantomas tendinosos.
En todas estas situaciones se recomienda realizar un estudio completo del perfil lipídico y hacer una evaluación de los componentes del índice DLCN. En las personas con valores de alta probabilidad o certeza diagnóstica clínica se recomienda el estudio genético mediante el Liponext.
Dada la elevada prevalencia de la alteración, probablemente superior a 1/50015, diversas sociedades científicas sugieren campañas de detección precoz dirigidas a toda la población. Estas consistirían en determinar las cifras de colesterol a los 8-9años de edad16, dado que en esta etapa de la vida es cuando las cifras de colesterol son más discriminativas entre los niños sanos y los afectados. Además, las guías clínicas de prevención cardiovascular recomiendan determinar las concentraciones de colesterol a partir de los 20años.
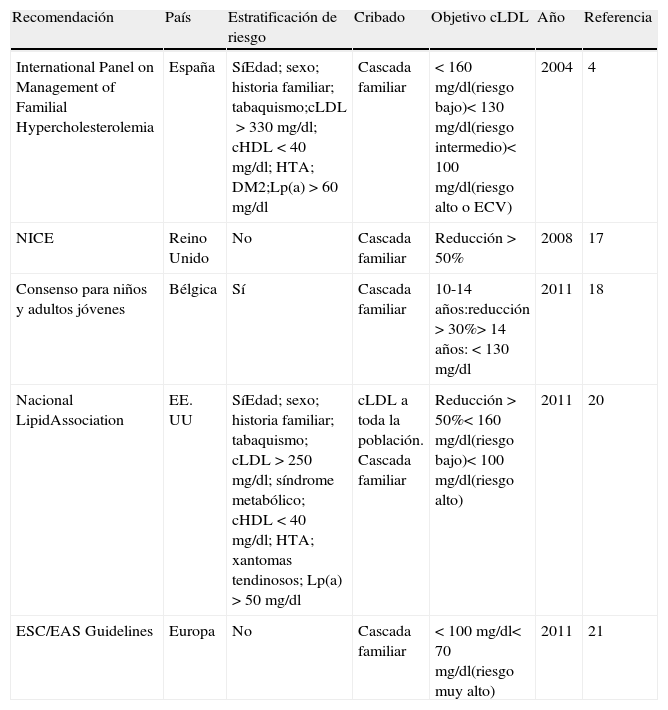
Principales documentos de consenso y guías clínicas sobre hipercolesterolemia familiarDesde la publicación a escala internacional de las primeras guías clínicas para el manejo de la HF ha pasado casi una década4. La Sociedad Española de Arteriosclerosis (SEA) tuvo el acierto de reunir a un grupo de expertos internacionales que elaboraron unas recomendaciones que han tenido una gran repercusión en el manejo de la enfermedad desde entonces. Aquellas primeras guías se han seguido de otras conforme el conocimiento de la enfermedad ha ido avanzando y la evidencia científica del beneficio del tratamiento ha ido aumentando. En este apartado resumimos las recomendaciones que, con repercusión internacional, se han publicado desde entonces (tabla 4).
Resumen de las principales recomendaciones internacionales
| Recomendación | País | Estratificación de riesgo | Cribado | Objetivo cLDL | Año | Referencia |
| International Panel on Management of Familial Hypercholesterolemia | España | SíEdad; sexo; historia familiar; tabaquismo;cLDL>330mg/dl; cHDL<40 mg/dl; HTA; DM2;Lp(a)>60 mg/dl | Cascada familiar | <160 mg/dl(riesgo bajo)<130 mg/dl(riesgo intermedio)<100 mg/dl(riesgo alto o ECV) | 2004 | 4 |
| NICE | Reino Unido | No | Cascada familiar | Reducción >50% | 2008 | 17 |
| Consenso para niños y adultos jóvenes | Bélgica | Sí | Cascada familiar | 10-14 años:reducción >30%>14años: <130mg/dl | 2011 | 18 |
| Nacional LipidAssociation | EE.UU | SíEdad; sexo; historia familiar; tabaquismo; cLDL>250 mg/dl; síndrome metabólico; cHDL<40 mg/dl; HTA; xantomas tendinosos; Lp(a)>50 mg/dl | cLDL a toda la población. Cascada familiar | Reducción >50%<160 mg/dl(riesgo bajo)<100 mg/dl(riesgo alto) | 2011 | 20 |
| ESC/EAS Guidelines | Europa | No | Cascada familiar | <100 mg/dl<70 mg/dl(riesgo muy alto) | 2011 | 21 |
cHDL: colesterol de las lipoproteínas de alta densidad; cLDL: colesterol de las lipoproteínas de baja densidad; DM2: diabetes mellitus tipo2; ECV: enfermedad cardiovascular; HTA: hipertensión arterial; Lp(a): lipoproteína(a).
Promovido por la SEA y con el apoyo de la Sociedad Española de Cardiología y la Fundación Española de Hipercolesterolemia Familiar, un panel de expertos internacionales propusieron las primeras recomendaciones a nivel mundial para el diagnóstico y el tratamiento de la hipercolesterolemia familiar. Entre las principales recomendaciones del documento se encontraban la necesidad de un diagnóstico precoz de la enfermedad; una estrategia de detección basada en la búsqueda de familiares afectados una vez identificado un probando; la estratificación del riesgo en los sujetos heterocigotos de acuerdo con la presencia de otros factores de riesgo; la detección precoz de la arteriosclerosis en fase preclínica; el establecimiento de 3objetivos terapéuticos de cLDL según el riesgo basal, y una estrategia terapéutica basada en un estilo de vida cardiosaludable y tratamiento farmacológico escalonado con estatinas potentes como primera opción, seguido de dosis máximas de estatinas potentes con ezetimiba o resinas de intercambio iónico como segundo nivel de tratamiento farmacológico. La aféresis de LDL se recomienda cuando después del tratamiento farmacológico el cLDL es >200mg/dl en presencia de enfermedad coronaria, o por encima de 300mg/dl en ausencia de ella4.
Otra recomendación novedosa de este documento fue la sugerencia de iniciar precozmente el tratamiento farmacológico en la HF, de acuerdo con la edad, el sexo y el riesgo basal de los sujetos.
Este documento sigue vigente en la actualidad y, como veremos a continuación, las recomendaciones posteriores avalan las realizadas por la SEA en aquel momento.
National Institute for Health and Clinical Excellence del Reino UnidoEn el año 2008 se publicaron las guías clínicas británicas NICE para HF. En ellas se recomienda el diagnóstico clínico basado en los criterios del registro británico de Simon Broome. En ellos se da un peso muy elevado a la presencia de enfermedad coronaria prematura y/o xantomas, y, sin embargo, las concentraciones diagnósticas de cLDL son relativamente bajas. Curiosamente, no recomienda un objetivo de cLDL; en su lugar aconsejan reducir más del 50% la concentración de cLDL. Tampoco se hace una estratificación del riesgo basal para instaurar un tratamiento, y recomiendan iniciar la terapia farmacológica a partir de los 10años de edad17.
Consenso Belga para el Tratamiento de la Hipercolesterolemia Familiar en Niños y Adultos JóvenesPublicado en el año 2011, está muy centrado en el diagnóstico y el tratamiento de los niños18. Con respecto a los adultos, dice textualmente: «El beneficio, objetivos y tratamiento de los adultos con HF heterocigota está ya bien definido», y como referencia citan al consenso del año 2004 de la SEA, que consideran de total actualidad. Para los niños recomiendan el diagnóstico a partir de los 2años en presencia de un progenitor afectado, con la idea de establecer un tratamiento dietético pobre en grasa saturada a partir de esa edad. El tratamiento farmacológico lo consideran a partir de los 10años cuando el cLDL sea >190mg/dl, o por encima de 160mg/dl en presencia de historia de enfermedad coronaria prematura en la familia o factores de riesgo cardiovascular. El objetivo del tratamiento lo establecen en reducir el cLDL>30% entre los 10 y los 14años, y lograr cLDL<130mg/dl a partir de los 14años18.
National Lipid Association Expert Panel on Familial HypercholesterolemiaEsta asociación de lipidólogos de Estados Unidos publicó un número monográfico de la revista Journal of Clinical Lipidolgy en 2011 dedicado a la HF19. En dicho número se presentan las principales recomendaciones de esta asociación e incluye capítulos sobre diagnóstico, cribado y tratamiento, tanto para niños como para adultos. Recomiendan preferentemente el diagnóstico clínico basado en las concentraciones de cLDL ajustadas a la edad. Se inclinan por un cribado general a toda la población con concentraciones de cLDL>190mg/dl en la edad adulta, o por encima de 160mg/dl en niños. Con respecto al tratamiento, recomiendan iniciar el tratamiento farmacológico cuando el cLDL es >190mg/dl, con objetivos terapéuticos diferentes en dependencia de los factores de riesgo del sujeto20, a semejanza de la recomendación promovida por la SEA.
ESC/EAS Guidelines for the Management of DyslipidaemiasLas sociedades europeas de Cardiología (ESC) y Arteriosclerosis (EAS), en su documento de manejo clínico de las dislipidemias publicado en 2011, dedica una sección a la HF con directrices específicas para esta enfermedad21. En ellas recomienda el diagnóstico tanto clínico como genético de la enfermedad, así como el cribado basado en la extensión del estudio familiar, y fija unos objetivos muy agresivos, ya que considera a todos los sujetos afectos de HF heterocigota como sujetos de alto riesgo vascular y recomienda objetivos de cLDL<100mg/dl, y <70mg/dl cuando el HF es un sujeto de muy alto riesgo por tener enfermedad coronaria, diabetes u otro equivalente coronario.
Gracias a los esfuerzos de la comunidad científica y de muchas asociaciones médicas y de pacientes, el diagnóstico y el tratamiento con hipolipemiantes de los sujetos con HF ha mejorado sustancialmente en los últimos años. El pronóstico de los sujetos heterocigotos tratados, con unas u otras recomendaciones, ha conseguido reducir drásticamente la enfermedad cardiovascular en los pacientes diagnosticados en muchos países occidentales22. El reto fundamental en los próximos años es expandir el diagnóstico en nuestro medio y que el tratamiento prolongado sea la norma en este colectivo de pacientes paradigma de la prevención cardiovascular y de la intervención médica coste-efectiva23.
Tratamiento farmacológicoHa quedado patente en los apartados anteriores que los pacientes con HF heterocigota son un grupo poblacional de alto riesgo cardiovascular, con concentraciones plasmáticas de cLDL muy elevadas y con amplia variabilidad familiar dependiendo de la clase de mutación en el gen del LDLr. El tratamiento precoz de este grupo poblacional es beneficioso y requiere estrategias intensivas de intervención, por lo que es recomendable que su control se realice en las unidades de lípidos y riesgo vascular. El objetivo terapéutico para disminuir el riesgo cardiovascular en los pacientes con HF heterocigota es alcanzar una concentración de cLDL<100mg/dl, o por lo menos una reducción del 50%, por lo que será preciso utilizar estatinas potentes, con inhibidores de la absorción de colesterol (ezetimiba), y en su caso con resinas (preferentemente colesevelam) a altas dosis, tratamiento combinado y, en ocasiones, aféresis de LDL20.
A pesar de que las modificaciones higiénico-dietéticas no suelen ser suficientes para alcanzar las concentraciones deseables de cLDL, el estilo de vida saludable sigue siendo un pilar fundamental en el tratamiento de la HF, con muchos beneficios más allá del descenso del cLDL. La modificación del estilo de vida comprende una dieta saludable, la reducción del peso corporal cuando hay sobrepeso/obesidad, no fumar y una actividad física moderada21.
La dieta recomendada incluye una menor ingesta de grasas saturadas y colesterol (grasa total 25-35% de las calorías totales, grasas saturadas menos del 7% de las calorías totales, colesterol menos de 200mg/día), el uso de esteroles vegetales (1,5-2g/día) y de fibra soluble (10-20g/día)24.
Por lo que hace referencia al tratamiento farmacológico, las estatinas son los fármacos de elección. Se deberán administrar las estatinas más eficaces a las dosis más altas toleradas. Los datos observacionales de 2grandes cohortes europeas de pacientes HF confirman la eficacia de las estatinas en la HF heterocigota25,26. Las estatinas son seguras, bien toleradas y los efectos adversos son similares a los observados en la población dislipidémica que recibe dicho tratamiento, particularmente el aumento de las enzimas hepáticas y la miopatía.
Los pacientes que no toleran estatinas potentes a altas dosis deberán recibir estatinas de baja/moderada potencia a dosis bajas o combinación con otros hipolipemiantes. Las estatinas de baja potencia presentan un menor riesgo de miotoxicidad27, pero no permiten alcanzar la reducción de más del 50% en cLDL recomendada en estos pacientes20. La medicación hipolipemiante en estos casos incluye la ezetimiba y los secuestradores de ácidos biliares. Incluso los pacientes que toleran las dosis máximas de las estatinas más potentes requieren a menudo una combinación de uno o más fármacos para alcanzar los objetivos recomendados. Dado el perfil de tolerabilidad y seguridad de la ezetimiba, es razonable considerar, como una segunda etapa de tratamiento, la adición de este inhibidor de la absorción del colesterol intestinal a la dosis máxima de estatinas en pacientes con HF que requieren la intensificación del tratamiento. Con el tratamiento de coadministración se consigue una reducción adicional del 15 al 25% en cLDL28. El tercer paso es la terapia triple, consistente en una estatina con ezetimiba y un secuestrador de los ácidos biliares; en este sentido, la adición de colesevelam a una estatina y ezetimiba disminuye las concentraciones de cLDL un 12% adicional29.
Las resinas tienen importantes interacciones con otros fármacos, por lo que deben ser administradas 4h antes o 1h después de otros fármacos. El colesevelam es mejor tolerado y presenta menos interacciones, especialmente con las estatinas.
En el mundo real, una gran proporción de los pacientes con HF no alcanzan los objetivos terapéuticos y, por consiguiente, persiste un elevado riesgo de enfermedad cardiovascular30. Así, por ejemplo, en un gran estudio transversal llevado a cabo en los Países Bajos31, casi todos los pacientes con HF heterocigota (96%) estaban en tratamiento con estatinas. Solo el 21% de los pacientes alcanzaron el objetivo de cLDL por debajo de 100mg/dl (menos de 2,5mmol/l) y cerca del 5% de los pacientes todavía tenía una concentración de cLDL>200mg/dl. Entre los que no habían logrado el objetivo terapéutico, el 27% estaban en terapia de combinación de dosis máxima de estatina y ezetimiba. Estos resultados subrayan la necesidad de disponer de nuevas terapias farmacológicas reductoras de LDL.
Aféresis de lipoproteínas de baja densidadLas guías clínicas establecen la indicación de aféresis de LDL en HF homocigota y en los pacientes con HF heterocigota y enfermedad cardiaca coronaria establecida, cuando tras un tratamiento farmacológico a dosis máximas la concentración de cLDL es >160mg/dl o, en pacientes con HF sin enfermedad cardiovascular, >200mg/dl32. Los diferentes métodos de aféresis de LDL consiguen objetivos similares, con una reducción del cLDL del 60-70% sobre las cifras previas a la aféresis, que se recuperan progresivamente. Por ello, los pacientes con HF precisan repetir el procedimiento, cada 15días los heterocigotos y cada 7-10días los homocigotos. Además, con la aféresis de LDL, la Lp(a) disminuye un 58% y los triglicéridos un 27%, sin diferencias significativas entre los distintos métodos33. El efecto beneficioso demostrado a largo plazo por la aféresis de LDL sobre la enfermedad cardiaca coronaria y vascular periférica se atribuye a la mejoría del perfil lipoproteico, aunque probablemente intervengan otros mecanismos, como cambios en las moléculas de adhesión y marcadores de inflamación. Por lo general, el procedimiento es bien tolerado, con una frecuencia de incidencias clínicas inferior al 5%. La mayor parte de los efectos secundarios son leves, como hipotensión arterial transitoria o problemas en el acceso venoso. Su principal limitación está relacionada en el coste económico y la baja disponibilidad.
Situaciones especialesNiñosComo norma general, el control de la dislipidemia en los niños debe centrarse en la dieta y el tratamiento de los trastornos metabólicos subyacentes, y exclusivamente en aquellos con HF debe considerarse la posibilidad de tratamiento con fármacos hipolipemiantes. Las recomendaciones dietéticas tienen que ser consistentes con una buena alimentación. A pesar de que no disponemos de estudios que hayan evaluado de forma adecuada el efecto de las dietas restringidas en grasa y colesterol en niños con HF heterocigota34, estas se acompañan de modestas caídas en los niveles de cLDL (0,13mmol/l a los 3años de seguimiento)35. Por otra parte, hay que destacar el efecto beneficioso de los esteroles vegetales en esta población, con reducciones en cLDL similares a la población adulta y con una excelente tolerancia y fácil cumplimiento36,37.
En base a las evidencias disponibles38, las últimas guías clínicas18,37,39 recomiendan no iniciar el tratamiento farmacológico antes de los 10años de edad, siendo las estatinas el fármaco hipolipemiante de primera línea. Sin embargo, en los niños con HF homocigota o heterocigota severa, con una concentración de cLDL>400mg/dl, en aquellos con enfermedad cardiovascular establecida o portadores de un trasplante cardiaco puede instaurarse el tratamiento a edades más tempranas40. Recientemente, Vuorio et al.41 han ilustrado el potencial beneficio clínico de iniciar la terapia con estatinas a la edad de 10años, calculando, en base a la analogía de paquetes/año en el tabaquismo, el cLDL acumulado como la suma de los niveles de cLDL multiplicado por la edad en años. A pesar de que el nivel de evidencia es bajo, el objetivo terapéutico recomendado en la población infanto-juvenil con HF heterocigota es alcanzar un cLDL≤130mg/dl o una reducción de al menos el 30% del valor basal entre los 10 y los 14años.
En un estudio aleatorizado, doble ciego y controlado con placebo en niños de 10 a 17años de edad en el que se coadministró ezetimiba con simvastatina, los pacientes mostraron una reducción adicional del 15% en los niveles de cLDL en comparación con la administración de estatinas en monoterapia42.
Su uso como monoterapia aún no se ha investigado a fondo en niños, aunque un estudio retrospectivo sugiere que puede ser eficaz, mostrando un descenso en cLDL del 25%43. Este fármaco representa una terapia potencialmente importante en los pacientes jóvenes que requieren importantes descensos en cLDL, ya que por su perfil de seguridad no aumentan los efectos secundarios del tratamiento con estatinas.
No sería justo dejar de mencionar que la mayoría de los estudios clínicos realizados con estatinas en los niños han sido de corta duración y, por tanto, no disponemos de datos de seguridad a largo plazo en pacientes que requerirán tratamiento farmacológico hipolipemiante de por vida. A pesar de ello, es poco probable que se lleven a cabo ensayos a largo plazo en niños y adolescentes, por lo que los resultados de los estudios observacionales pueden ser de gran utilidad16. Así, en un estudio observacional, los 185niños y adolescentes con HF heterocigota que fueron tratados con pravastatina y seguidos durante un máximo de 7años no mostraron efectos en el crecimiento ni en la maduración sexual, siendo la prevalencia global de efectos adversos del 13%, la mayoría leves44.
Existen estudios de intervención en los que se ha evaluado el efecto de diversas estatinas en niños con HF sobre el grosor íntima-media y la función endotelial. Dos metaanálisis muestran un efecto similar al observado en los adultos, y posteriormente se han publicado los datos de pravastatina 20mg, fluvastatina 80mg y rosuvastatina 5, 10 y 20mg. La Food and Drug Administration (FDA) y la Agencia Europea de Medicamentos (EMEA) han aprobado el uso de simvastatina, pravastatina, fluvastatina, atorvastatina y rosuvastatina en niños a partir de los 10años.
En la adolescencia es frecuente el uso de retinoides para el tratamiento del acné. La terapia no está contraindicada si es por períodos breves, si bien deberá suspenderse el tratamiento concomitante con estatinas.
Mujeres en edad fértil. Embarazo y lactanciaLas mujeres con HF heterocigota deberán recibir asesoramiento sobre los métodos anticonceptivos y las instrucciones para suspender los fármacos hipolipemiantes, como las estatinas y la ezetimiba, al menos 4semanas antes de dejar la anticoncepción, informándolas de que no deben usarlos durante el embarazo ni en la lactancia. En caso de embarazo no deseado, una mujer con HF heterocigota debe suspender las estatinas y la ezetimiba inmediatamente y consultar con su médico19. Un registro noruego de 2.319nacimientos en 1.093mujeres con HF heterocigota demuestra que la frecuencia de parto prematuro, recién nacidos de bajo peso o malformaciones congénitas fue similar a la de la población general de mujeres en edad fértil45.
Los únicos fármacos hipocolesterolemiantes que pueden ser considerados durante el embarazo son las resinas de intercambio o secuestradores de ácidos biliares, debido a que estos medicamentos no pasan a la circulación sistémica. Las resinas reducen un 15% la concentración de cLDL y, de las disponibles en la actualidad, el colesevelam es la mejor tolerada46. Dado que las resinas tienen la capacidad de aumentar la síntesis de triglicéridos, se debe tener precaución en las pacientes con trigliceridemia ≥300mg/dl, estando contraindicado en aquellas con niveles ≥400mg/dl, debido a un mayor riesgo de pancreatitis.
Habrá que considerar la aféresis de LDL durante el embarazo en las pacientes con HF heterocigota y enfermedad cardiovascular establecida o si son homocigotas.
La suspensión del tratamiento con estatinas y ezetimiba durante el embarazo debe prolongarse durante la lactancia materna, dado que las estatinas se eliminan a través de esta, por lo que debería desaconsejarse la lactancia materna o acortarla al mínimo necesario.
Diabetes mellitusDe acuerdo con la reciente guía del 5th Joint Task Force, los pacientes con diabetes tipo2 y un factor de riesgo o lesión de órgano diana o microalbuminuria son considerados de muy alto riesgo cardiovascular47. Por tanto, un paciente con HF heterocigota y diabetes será un paciente de muy alto riesgo, y el objetivo terapéutico a alcanzar en esta situación será un cLDL<70mg/dl.
Prevención secundariaDe acuerdo con la reciente guía del 5th Joint Task Force, los pacientes con enfermedad cardiovascular conocida, clínica o subclínica, son considerados de muy alto riesgo cardiovascular47. Por tanto, un paciente con HF heterocigota y enfermedad cardiovascular será considerado de muy alto riesgo y el objetivo terapéutico a alcanzar en esta situación será un cLDL<70mg/dl (1,8mmol/l) o conseguir, por lo menos, una reducción superior al 50% si el objetivo no es alcanzable.
Tanto en pacientes con HF y diabetes como cardiopatía isquémica será necesario administrar terapia hipolipemiante combinada con 2 o 3fármacos, incluyendo estatinas, ezetimiba y resinas.
Recomendaciones para la detección, el diagnóstico y el tratamiento de la hipercolesterolemia familiarDetección en la infanciaDetección mediante búsqueda generalizada. Diversas sociedades científicas abogan por la determinación de colesterol en niños y niñas a la edad de 8-10años, dado que es el momento de mayor discriminación de las cifras de colesterol entre afectados y no afectados. Consideramos que sería una política apropiada, pero debe ser apoyada oficialmente por las autoridades sanitarias.
En todo niño con cifras de colesterol total >200mg/dl se determinará el cLDL y se recomienda evaluar la posibilidad de HF en niños con cLDL>160mg/dl, especialmente si existen antecedentes familiares de hiperlipidemia.
Detección en la edad adultaToda persona debería conocer sus cifras de colesterol a partir de los 20años. Con concentraciones de colesterol total >250mg/dl se debe determinar el cLDL, y se recomienda evaluar el diagnóstico de HF si el cLDL>190mg/dl.
Detección por cascada diagnósticaEstá indicada la práctica del estudio genético en todos los familiares de primer grado de los pacientes diagnosticados de HF con mutación positiva, independientemente de sus cifras de colesterol.
En las familias en las que se ha establecido el diagnóstico de certeza o probabilidad, aunque no se haya detectado una mutación responsable, está indicado el cribado en cascada mediante determinaciones de cLDL en los familiares de primer grado.
Cribado en cascada inversaToda persona debería conocer sus cifras de colesterol a partir de los 20años. Con concentraciones de colesterol total >250mg/dl se debe determinar el cLDL y se recomienda evaluar el diagnóstico de HF si el cLDL>190mg/dl.
Está indicada la determinación de cLDL en todos los familiares de primer grado de los niños y niñas en los que se hayan detectado cifras de LDL>135mg/dl o se haya establecido el diagnóstico genético.
Detección oportunísticaDescartar HF mediante determinación de colesterol y aplicación del índice DLCN en:
- 1.
Familiares de primer grado de pacientes diagnosticados de HF.
- 2.
Pacientes con historia familiar de cardiopatía isquémica precoz.
- 3.
Pacientes con historia personal de cardiopatía isquémica precoz.
- 4.
Cifras de cLDL>190mg/dl, o >160mg/dl en niños y adolescentes.
- 5.
Detección de arco corneal antes de los 45años.
- 6.
Presencia de xantomas tendinosos. La práctica de ecografía del tendón de Aquiles permite detectar la presencia de xantomas tendinosos no percibidos en la exploración física.
- 1.
El diagnóstico se basa en el índice DLCN y el estudio genético.
- 2.
Un índice DLCN≥8 indica un diagnóstico de certeza clínica y los pacientes deben ser controlados y tratados como HF, aunque no se detecte la mutación.
- 3.
El estudio genético está indicado en pacientes con DLCN>6.
- 4.
El estudio genético está indicado en todos los familiares de primer grado de un paciente diagnosticado de HF con mutación positiva.
- 5.
Se recomienda la determinación del índice tobillo-brazo y del grosor íntima-media en el momento del diagnóstico y, si es normal, al menos cada 5años.
- 6.
Se deberá determinar la Lp(a) en todos los pacientes con HF.
- 7.
Se estudiará la presencia de otros factores de riesgo vascular, específicamente los antecedentes familiares de cardiopatía isquémica precoz, diabetes, hipertensión arterial y tabaquismo.
- 1.
Los objetivos terapéuticos de cLDL a alcanzar en los pacientes con HF son los siguientes:
- •
Niños: cLDL<130mg/dl, o reducción del 30% entre los 10 y los 14años.
- •
Adultos sin factores de riesgo adicionales: cLDL<115mg/dl.
- •
Adultos con al menos un factor de riesgo adicional: cLDL<100mg/dl.
- •
Adultos con diabetes o cardiopatía isquémica: cLDL<70mg/dl.
- •
- 2.
Todos los pacientes deben recibir las indicaciones correctas para adecuar su estilo de vida a hábitos cardiosaludables. La dieta de tipo mediterráneo con el uso de aceite de oliva como grasa culinaria y la ingesta diaria de frutos secos es recomendable. La adición de alimentos funcionales enriquecidos con fitosteroles colabora a disminuir el cLDL.
- 3.
Se debe tratar el hábito tabáquico enérgicamente para conseguir su abandono.
- 4.
Se estimulará la práctica de ejercicio físico, como mínimo caminar 30min al día.
- 5.
Desde el momento del diagnóstico, en los adultos se iniciará tratamiento con las estatinas más potentes, alcanzando en el menor tiempo posible las dosis más altas: rosuvastatina 20-40mg; atorvastatina 80mg. Si a los 2meses no se han alcanzado los objetivos, se coadministrará ezetimiba.
- 6.
En pacientes en los que no se consiga el objetivo terapéutico (cLDL<100mg/dl, o <70mg/dl si son diabéticos o con enfermedad cardiovascular) con 2fármacos, se recomienda añadir un tercero (resinas: preferentemente colesevelam 3,7g/día).
- 7.
El tratamiento farmacológico en los niños se iniciará a partir de los 10años con cLDL>190mg/dl, o cLDL>160 y <190mg/dl si hay factores de riesgo adicionales, especialmente con antecedentes familiares de enfermedad cardiovascular precoz, utilizando las estatinas, que se han mostrado eficaces y seguras en estas edades. La ezetimiba puede ser también utilizada en combinación.
- 8.
Se realizarán controles analíticos anuales una vez controlados los pacientes.
- 9.
Se recomienda aféresis de LDL en pacientes HF con cardiopatía isquémica que, a pesar de seguir tratamiento farmacológico máximo, mantengan cifras de cLDL>160mg/dl o en pacientes sin cardiopatía isquémica con cLDL>200mg/dl.
- 10.
A las mujeres en edad fértil se les debe asesorar sobre las medidas anticonceptivas y la necesidad de abandonar el tratamiento al menos un mes antes del embarazo.
- 11.
Las pacientes embarazadas han de suspender el tratamiento con estatinas y ezetimiba, si es posible, un mes antes del embarazo. En pacientes con riesgo muy elevado puede valorarse el tratamiento con resinas. Esta pauta terapéutica debe mantenerse durante la lactancia materna, que debería desaconsejarse o acortarse al mínimo posible.
Ferrer Internacional (Ferrer Grupo) ha dado soporte al comité científico para desarrollar el presente documento de consenso, no participando ni en el diseño, ni en el análisis de datos, ni en la redacción del presente artículo.