


Tradicionalmente se ha pensado que las pruebas bioquímicas del metabolismo fosfocálcico permiten diferenciar el hiperparatiroidismo primario (HPT1) y la hipercalcemia hipocalciúrica familiar (HHF) pero hay casos de difícil diagnóstico incluso para clínicos experimentados.
Nos planteamos como objetivo evaluar la validez de las pruebas diagnósticas de la HHF así como la correcta indicación del estudio genético.
Pacientes y métodosHemos realizado un estudio descriptivo de 2 familias con hipercalcemia y sospecha de HHF de características atípicas.
En orina de 24h hemos valorado los índices de excreción urinaria de calcio (eliminación de calcio [CE], cociente calcio/creatinina [CR] y cociente aclaramiento de calcio/aclaramiento de creatinina [CCCR]), junto con las concentraciones séricas de PTH y 25 hidroxivitamina D. A los casos índices se les realizó el estudio genético.
ResultadosUna paciente presentó hipercalciuria franca y persistente con valores más concordantes con HPT1 que de HHF si consideramos, como proponen las guías, un CCCR inferior a 0,01 como indicativo de HHF y superior a 0,02 como HPT1. Al caso índice de la segunda familia se le extirpó un adenoma de paratiroides. En ambos casos índice, encontramos la misma mutación c. 164C>T (p.Pro55Leu) en el exón 2 en heterocigosis descrita como responsable de HHF.
ConclusionesEl diagnóstico definitivo de HHF en las guías clínicas actuales requiere confirmación genética, lo cual ha permitido en nuestro caso la detección de 2 familias con HHF y características clínicas atípicas. En nuestra opinión el uso racional de estas pruebas ante la sospecha de HFF puede evitar intervenciones quirúrgicas innecesarias y gastos excesivos en su monitorización.
Biochemical tests related to calcium and phosphorus metabolism have traditionally been considered as a reliable tool to differentiate familial hypocalciuric hypercalcemia (FHH) from primary hyperparathyroidism (PHPT). However, diagnosis may sometimes be difficult even for experienced clinicians. Our objective was to assess the accuracy of diagnostic tests in FHH and the circumstances in which genetic studies are required.
Patients and methodsA descriptive study was conducted of two families with hypercalcemia and suspected atypical FHH. Urinary calcium excretion was measured in 24-hour urine using different tests (calcium excretion (CE), urinary calcium/creatinine clearance ratio (UCCR)), and serum PTH and 25-hydroxyvitamin D levels were tested. Index cases underwent genetic study.
ResultsOne patient from the first family showed overt, persistent hypercalciuria with values more consistent with PHPT than with FHH if we consider, as proposed by guidelines, a UCCR lower than 0.01 as diagnostic of FHH and a value higher than 0.02 as diagnostic of PHPT. The index case of the second family underwent surgery for a parathyroid adenoma. Both cases had a mutation c. 164C>T (Pro55Leu) in exon 2 in heterozygosis.
ConclusionsAccording to current clinical guidelines, definitive diagnosis of FHH requires genetic confirmation, which allowed in our case for detection of two families with FHH and atypical clinical presentations. We think that rational use of genetic tests may avoid unnecessary surgery and excess monitoring costs.
Las hipercalcemias familiares no son infrecuentes en las consultas de endocrinología. Son trastornos hereditarios que se transmiten casi siempre de forma autosómica dominante1,2 y abarcan un espectro de enfermedades que incluyen la neoplasia endocrina múltiple tipos 1 y 2 (MEN 1 y 2), el hiperparatiroidismo asociado a tumor mandibular, el hiperparatiroidismo familiar aislado y la hipercalcemia hipocalciurica familiar (HHF). Distinguir los 5 síndromes es a veces difícil pero tiene profundas implicaciones para el paciente y su familia.
La disponibilidad de pruebas genéticas específicas ha mejorado la precisión diagnóstica y simplificado la monitorización pero tienen un coste económico elevado, muchas veces no son fácilmente accesibles y por tanto requieren un uso racional.
Las mutaciones inactivantes del gen del receptor sensible al calcio (CaSR), localizado en el brazo largo del cromosoma 3, pueden afectar a un solo alelo (heterocigosis) en cuyo caso se genera el fenotipo característico de HHF o a los 2 alelos (homocigosis o heterocigosis compuesta si no hay consanguinidad) originando un hiperparatiroidismo neonatal grave, de forma que el grado de defecto génico explica la gran disparidad de la presentación fenotípica3.
En la HHF se han descrito unas 200 mutaciones diferentes4 cuyo resultado final es la disminución del número de receptores funcionantes en las células paratiroideas y riñón, lo que las hace parcialmente resistentes al calcio, de forma que se requieren concentraciones elevadas de éste para disminuir la PTH y se reabsorbe calcio y magnesio en el riñón aun en presencia de hipercalcemia. Cursa por tanto con hipercalcemia persistente, hipocalciuria absoluta (típicamente menor de 200mg/día) o relativa al calcio plasmático (cociente aclaramiento de calcio/aclaramiento de creatinina –CCCR– menor de 0,01) y concentraciones normales de PTH, aunque el 20% de los pacientes tienen concentraciones elevadas de PTH5,6. Es un trastorno considerado benigno, asintomático y no parece acortar la esperanza de vida pero en algunos adultos se ha descrito condrocalcinosis y puede conferir una mayor susceptibilidad a presentar pancreatitis7. Es importante distinguir esta entidad del hiperparatiroidismo (HPT) primario, lo que suele ser fácil en los casos típicos de ambas entidades, porque en la HHF la cirugía es innecesaria e ineficaz.
Con el estudio genético el espectro clínico de las mutaciones del receptor sensible al calcio se está ampliando, describiéndose casos que cursan con hipercalciuria, litiasis8 y, excepcionalmente, con adenomas de paratiroides en pacientes con HHF9.
Material y métodosSe trata de un estudio descriptivo de 2 familias procedentes de la misma área geográfica pero sin antecedentes familiares comunes conocidos con hipercalcemia, a lo largo de años de seguimiento, en las que recientemente mediante estudio genético hemos confirmado que presentan HHF, a pesar de los datos clínicos y bioquímicos discordantes que presentaban.
Con 4 miembros afectados, el caso índice de la primera familia es una mujer de 35 años, 54,4kg de peso, 158cm de talla e índice de masa corporal (IMC) de 21kg/m2 a la que en el estudio de una cefalea se le detecta un calcio elevado, 11,1mg/dl, fósforo ligeramente disminuido, 2,4mg/dl, con magnesio y creatinina normales. Calciuria de 24h de 272mg, reabsorción tubular de fosfato (RTP) 70%, cociente aclaramiento de calcio/aclaramiento de creatinina (CCCR) 0,0137. Concentraciones de PTH intacta elevadas 110 pg/ml y déficit de 25 hidroxivitamina D 24,8 nmol/l. La ecografía de paratiroides y gammagrafía con Tc-sestamibi no revelaron agrandamiento de glándulas paratiroides, adenomas o tumores ectópicos. La ecografía abdominal fue normal así como la densitometría ósea de columna con Tscore: –0,02 y Zscore: 0,50.
Al estudiar la familia descubrimos que su padre de 70 años de edad tenía una hipercalcemia asintomática al igual que sus 2 hijos de18 y 12 años. El hijo mayor comenzó poco después del diagnóstico con episodios repetidos de cólicos nefríticos y hematuria macroscópica pero sin expulsión de cálculos, no visibles en la radiología abdominal convencional, siendo atendido de urgencias en nuestro hospital por ese motivo en 2004, 2008 y 2010.
En la segunda familia, el caso índice es un varón de 39 años con 92kg de peso y una talla 173cm (IMC: 30,7kg/m2) con hipercalcemia descubierta en el estudio de una hiperlipemia. Al estudiarlo, encontramos que 2 de 4 hermanos, incluido el caso índice, al igual que la madre y un tío, tenían hipercalcemia. En la analítica inicial tenía un calcio de 11,7mg/dl, fósforo de 2,74mg/dl y magnesio de 2,19mg/dl. Calciuria: 82mg/24h, CCCR: 0,003, fosfaturia: 1.254mg /24h y RTP de 80%. La PTH intacta estaba elevada 72 pg/ml y las concentraciones de 25 hidroxivitamina D disminuidas, 40 nmol/l (VN: 50-250). La densitometría ósea mostraba osteopenia de columna (Tscore: –1,30 y Zscore: –1,20). Ecografía tiroidea normal pero en la gammagrafía con Tc-sestamibi se apreció retención patológica del radiofármaco sobre porción inferior del lóbulo izquierdo tiroideo, compatible con adenoma de paratiroides sobre glándula paratiroides inferior izquierda.
El calcio, fósforo, magnesio, fosfatasa alcalina y creatinina se determinaron en ayunas por fotometría con un aparato Cobas c711 Hitache (Roche Diagnostics).
Se analizó la orina de 24h con calciuria, fosfaturia y reabsorción tubular de fosfatos bajo una dieta sin restricciones, descartando que los pacientes tomaran diuréticos. En ella se valoraron los 3 índices más utilizados de la excreción urinaria de calcio, considerando hipercalciuria si la eliminación de calcio (CE) es superior a 4mg/kg/24h o mayor de 240mg en mujeres y 300 en hombres, el cociente calcio/creatinina (CR), si mayor de 0,2mg/mg y el CCCR (calcio en orina de 24h x creatinina sérica/calcio sérico x creatinina en orina), considerando hipocalciuria si éste es inferior a 0,01.
Se determinó la PTH intacta por quimioluminiscencia (Inmulite 2000. Siemens, límites de normalidad entre 11 y 67 pg/ml) y la 25 hidroxivitamina D también por quimioluminiscencia con concentraciones de referencia entre 50 y 250 nmol/l (consideramos concentraciones de deficiencia grave las inferiores a 25, insuficiencia entre 25-50 y subóptimas entre 50 y 75 nmol/l en Andalucía10).
A los casos índices de cada familia se les solicitó consentimiento para realizar estudio de genética molecular para el gen del receptor del calcio, estudio que se realizó en el servicio de Bioquímica y Genética molecular del Hospital Clínico de Barcelona.
El ADN se extrajo a partir de leucocitos procedentes de sangre total, mediante el QIAmp DNA (QIAGEN). Para el análisis del gen CaSR (Ensembl: ENSG00000036828) se amplificaron por PCR las regiones codificantes de los exones 2, 3, 4, 5, 6 y 7 mediante primers flanqueantes (para el diseño de los primers se utilizó http://frodo.wi.mit.edu/primer3/). Los productos de PCR se secuenciaron mediante el BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) y se purificaron utilizando el sistema de Millipore (96 well plates Multiscreen PCRu96 & Montage seq96). A continuación, los productos se analizaron en un ABI Prism Genetic Analyzer 3130.xl (Applied Biosystems).
ResultadosDe los 3 índices de excreción renal de calcio (CE, CR y CCCR), el CCCR es considerado el de elección por su mejor poder discriminativo. Las guías proponen un punto de corte para el CCCR inferior a 0,01 para el diagnóstico de HHF, lo que indica que más del 99% del calcio filtrado es reabsorbido a pesar de la presencia de hipercalcemia, y superior a 0,02 para el de HPT primario.
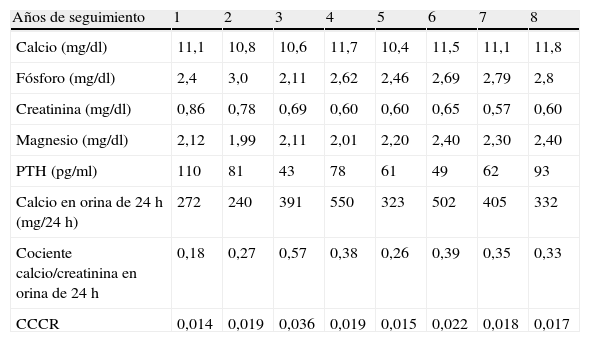
En la tabla 1 se recogen las variables descriptivas analíticas básicas en los 8 años de seguimiento del caso índice de la primera familia.
Variables descriptivas analíticas básicas del caso índice de la primera familia
| Años de seguimiento | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Calcio (mg/dl) | 11,1 | 10,8 | 10,6 | 11,7 | 10,4 | 11,5 | 11,1 | 11,8 |
| Fósforo (mg/dl) | 2,4 | 3,0 | 2,11 | 2,62 | 2,46 | 2,69 | 2,79 | 2,8 |
| Creatinina (mg/dl) | 0,86 | 0,78 | 0,69 | 0,60 | 0,60 | 0,65 | 0,57 | 0,60 |
| Magnesio (mg/dl) | 2,12 | 1,99 | 2,11 | 2,01 | 2,20 | 2,40 | 2,30 | 2,40 |
| PTH (pg/ml) | 110 | 81 | 43 | 78 | 61 | 49 | 62 | 93 |
| Calcio en orina de 24 h (mg/24 h) | 272 | 240 | 391 | 550 | 323 | 502 | 405 | 332 |
| Cociente calcio/creatinina en orina de 24 h | 0,18 | 0,27 | 0,57 | 0,38 | 0,26 | 0,39 | 0,35 | 0,33 |
| CCCR | 0,014 | 0,019 | 0,036 | 0,019 | 0,015 | 0,022 | 0,018 | 0,017 |
CCCR: cociente aclaramiento de calcio/aclaramiento de creatinina.
Como vemos, nuestra paciente, al valorar la eliminación de calcio en 24h, ha tenido franca y persistente hipercalciuria a lo largo de los años si consideramos la CE y CR. Al valorar los resultados del CCCR encontramos valores intermedios y algunos claramente indicativos de HPT primario más que de HHF.
Estas cifras de hipercalciuria además coinciden con un déficit asociado de vitamina D, cuya concentración modula la gravedad del cuadro, al provocar una disminución adicional de la calciuria y justificar muchas veces la elevación de la PTH que se observa en la HHF y que presentaba nuestra paciente.
A diferencia de ella, sus 2 hijos mantienen una hipercalcemia hipocalciúrica desde 1996 con CCCR inferior a 0,01 o marginalmente por encima, permaneciendo el pequeño asintomático y con valores de 25 hidroxivitamina D normales (80 nmol/l). El hijo mayor en el año 2005 tuvo 3 episodios de cólicos nefríticos que requirieron tratamiento en urgencias. Presentaba entonces calcemia de 11,7mg/dl, fosforemia de 3,17mg/dl, CE en orina de 24h, 248mg, CCCR 0,0123, PTH inapropiadamente elevada de 84 pg/ml y 25 hidroxivitamina D disminuida 49 nmol/l. Las ecografías abdominal y cervical y la gammagrafía con Tc-sestamibi fueron normales. Se realizó exploración quirúrgica con paratiroidectomía subtotal sin encontrarse alteraciones anatomopatológicas en las paratiroides, persistiendo la hipercalcemia después de la intervención. En la orina de 24h las cifras de oxalato y citrato son normales pero su excreción de ácido úrico está persistentemente elevada. (hasta 1.542mg /24h), por lo que podría pensarse en la presencia de litiasis úrica, pero hasta la fecha no ha expulsado cálculos susceptibles de analizar.
Ante los datos discordantes de esta familia solicitamos en 2010 estudio genético en el caso índice y se observó la mutación c.164C>T(Pro55Leu) en el exón 2 que está descrita como responsable de HHF.
En el caso índice de la segunda familia, considerando la edad del paciente, las concentraciones de calcio, su osteopenia y la captación de la gammagrafía, se decidió también la exploración quirúrgica del cuello, explicándonos la hipocalciuria por sus concentraciones bajas de vitamina D. En la cirugía se extirpa un adenoma de paratiroides inferior izquierdo, y se realiza monitorización intraoperatoria de la PTH que disminuyó de 72 pg/ml a 47 pg/ml poscirugía. Aunque al día siguiente a la cirugía el calcio total se normalizó, 9,8mg/dl, en los años posteriores de seguimiento en nuestras consultas el paciente sigue con hipercalcemia hipocalciurica –CCCR siempre inferior a 0,01–, a pesar de haberse normalizado las concentraciones de vitamina D tras el tratamiento. En el último control: calcio 11,5mg/dl, fósforo 1,96mg/dl, magnesio 2,3mg/dl, CCCR 0,009, 25 hidroxivitamina D 120 nmol/l, y PTH 34,8 pg/ml.
Los otros 3 miembros de la familia en los años de seguimiento permanecen asintomáticos y con CCCR igualmente inferiores a 0,01.
Por los datos discordantes de su historia, adenoma de paratiroides intervenido y datos familiares de hipercalcemia hipocalciúrica, recientemente le solicitamos estudio genético y también se observó la mutación c. 164C>T (Pro55Leu) en el exón 2 en heterocigosis que está descrita como responsable de HHF.
DiscusiónEn los últimos años, con la disponibilidad de pruebas genéticas, se han encontrado familias con mutaciones heterocigotas del gen CaSR que presentan cuadros clínicos característicos del hiperparatiroidismo como hipercalciuria, cálculos renales y más raramente adenomas de paratiroides. Hemos seguido a 2 familias, con 8 miembros afectados, durante años por sospecha de HHF que se ha confirmado recientemente mediante test genético y encontramos que 2 pacientes tenían una evolución discordante, cursando uno de ellos con hipercalcemia hipercalciúrica mantenida y el otro con adenoma de paratiroides.
Las hipercalcemias familiares, al tener un patrón hereditario autosómico dominante, se diagnostican en familias con varios miembros afectados, muchos de ellos jóvenes. En pacientes que presentan hipercalcemia asintomática con hipocalciuria relativa, especialmente si existen antecedentes de cirugía paratiroidea ineficaz, se debe investigar el calcio en sangre y orina en sus familiares en varias generaciones, aunque aparentemente no estén afectados. La determinación de calcio en sangre y orina en 3 miembros de una familia tiene menos falsos negativos que hacer el estudio genético en un caso índice. Si en un caso individual no se encuentran otros miembros afectados en la familia es muy improbable tenga una HHF11, aunque no lo descarta pues se han descrito familias con herencia recesiva2 o puede tener una mutación de novo. Hasta en un 30% de las familias no se observan mutaciones en el gen CaSR, lo cual puede ser debido en algunos casos a que las mutaciones están en otras regiones (promotor, intrones, etc.) o también a que sean otros los genes implicados.
El hecho de que no sean pruebas imprescindibles por la benignidad del cuadro, su precio, tasa alta de falsos negativos y hasta hace poco su difícil accesibilidad para los clínicos, limitan el uso sistemático del test genético en la HHF. Sin embargo, este puede evitar intervenciones quirúrgicas innecesarias y gastos económicos cuantiosos en los casos dudosos12.
La mayoría de HHF e HPT primario familiares no suelen plantear problemas de diagnóstico diferencial, pero ambas enfermedades tienen presentaciones atípicas difíciles de diferenciar. Así el 10% de los HPT primarios tienen PTH normal y el 15-20% de las HHF cursan con PTH elevada. Las guías12 proponen un punto de corte para el CCCR inferior a 0,01 para el diagnóstico de HHF y superior a 0,02 para el de HPT primario, que son ampliamente citados y aceptados13. Un estudio14 que ha evaluado recientemente el poder de discriminatorio de los 3 índices de excreción renal establece el punto de corte óptimo en 0,0115, pero puede haber solapamiento en la excreción urinaria de calcio entre los 2 cuadros.
Algunos HTP primarios tienen una excreción renal de calcio disminuida, incluso con CCCR inferior a 0,001, especialmente si tienen déficit añadido de vitamina D actualmente tan frecuente en España y Andalucía, que presenta caracteres epidémicos15–17, y pacientes con HHF pueden ser hipercalciúricos o más raramente presentar litiasis1,8 lo que provoca errores diagnósticos aun en manos de clínicos experimentados.
Un 20% de los FHH y hasta un 12% de HTP primarios se clasificarían mal y serían potencialmente mal tratados, incluso aumentando el punto de corte del CCCR de inferior a 0,01 a 0,0115, por ello se ha sugerido13 una estrategia diagnóstica en 2 pasos en pacientes dudosos. Primero determinar el CCCR estableciendo un valor de corte inferior a 0,020. Esto excluye a dos tercios de los HTP primarios que tendrán un valor superior e incluirá al 98% de los HHF. El siguiente paso sería determinar el gen CaSR en las familias con CCCR inferior a 0,020, lo cual separa las mutaciones (HHF) de los que no las tienen (HTP primario).
Una causa de error es no valorar las concentraciones de vitamina D, imprescindibles en estos pacientes, pues su déficit provoca una disminución en la absorción intestinal y en la excreción renal de calcio, un incremento de los niveles de PTH y la inactivación del receptor sensible al calcio18. Los pacientes con HTP primario parecen tener concentraciones más bajas de 25 hidroxivitamina D que los controles y la excreción renal de calcio de estos pacientes se correlaciona positivamente con estos niveles, alcanzando en deficitarios niveles típicos de HHF10,19.
Por otra parte, hay que considerar que la intensidad de la mutación del gen CaSR es variable y condiciona el grado de secreción de la PTH, la reabsorción tubular de calcio y la calcemia de un caso individual.
Nuestros 2 casos índices de ambas familias con HHF presentaban unas concentraciones disminuidas de 25 hidroxivitamina D. En uno de ellos el tratamiento sustitutivo con vitamina D disminuyó las concentraciones de PTH pero, como está descrito en pacientes con HHF20, no cambió la excreción renal de calcio, que permaneció muy baja (CCCR inferior a 0,01). La ausencia de un incremento mantenido de la PTH en los pacientes con HHF contribuye a su curso benigno habitual y a la ausencia de afectación ósea21.
Uno de los casos tiene hipercalciuria mantenida, a pesar de tener un déficit asociado de vitamina D, con cifras de CCCR superiores a 0,02 durante años de seguimiento, lo que es excepcional. Uno de sus hijos presenta cólicos nefríticos de repetición asociados a hipocalciuria y valores elevados de ácido úrico en orina.
Al menos un 9% de las hipercalcemias persistentes tras cirugías ineficaces22 se deben a pacientes no diagnosticados de HHF. El examen anatomopatológico de las glándulas paratiroideas muestra un leve agrandamiento o una ligera hiperplasia de dichas glándulas pero habitualmente no existe nodularidad. Presentamos un caso de HHF genéticamente probado, con gammagrafía Tc- sestamibi indicativa de adenoma de paratiroides inferior izquierdo que se confirmó quirúrgicamente. Esta técnica tiene una sensibilidad del 85-100% para localizar adenomas preoperatoriamente y una especificidad cercana al 100% si no hay afectación tiroidea coexistente como en nuestro caso23.
Se desconoce si la HHF es un factor de riesgo14 para el desarrollo posterior de HTP primario. La expresión de la proteína del CaSR está con frecuencia reducida en adenomas de pacientes con HTP primario24, por lo que se podría especular con que la pérdida total o parcial de la función del receptor puede estimular la proliferación de la célula paratiroidea. Recientemente se ha comunicado la asociación de mutaciones inactivantes homocigotas del CaSR con HTP primario por adenomas paratiroideos múltiples e hipercalcemia persistente grave tras cirugía, lo que sugiere que la pérdida de la función del receptor puede ocasionalmente provocar HTP primario en adultos25. En raros casos las 2 enfermedades pueden ocurrir juntas. En los últimos 10 años en Medline hemos encontrado 4 casos publicados de adenomas de paratiroides en pacientes con HHF genéticamente comprobados8,9,26,27. El HTP primario es una enfermedad frecuente por lo que podría ser una mera coincidencia, pero la presencia en una de estas familias de adenomas de paratiroides en diferentes generaciones no parece casual9.
El déficit de 25 hidroxivitamina D en HTP primario se asocia a una evolución más agresiva con mayor afectación ósea, por lo que las nuevas guías28 recomiendan su repleción.
Para algunos autores el déficit de vitamina D puede tener un papel en el desarrollo de adenomas de paratiroides e indican que su déficit crónico acelera su crecimiento9,19 y que la suplementación de vitamina D puede revertir este proceso actuando sobre el alelo normal de los pacientes con HHF, pero existen datos contradictorios29,30.
En resumen, la disponibilidad de estudios genéticos esta permitiendo expandir el espectro clínico de la HHF. Para las guías clínicas actuales11 el diagnóstico definitivo de HHF requiere estudio genético. Analizamos los protocolos actuales para cribado genético en pacientes con sospecha de HHF para su uso racional. Un test genético para la mutación del CaSR en los casos atípicos, como los que nosotros presentamos, puede evitar cirugías innecesarias y según nuestra opinión costes excesivos en el seguimiento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
