




Costa Rica es conocida por tener un índice de prevalencia relativamente alto de enfermedad de Wilson (EW). En el presente estudio se describe la presentación clínica y las características demográficas de los pacientes diagnosticados con EW en el Hospital Nacional de Niños de Costa Rica.
MétodosRevisión retrospectiva de los expedientes de todos los pacientes diagnosticados de EW desde enero de 1992 hasta marzo de 2006.
Resultados35 pacientes fueron diagnosticados de EW, y un 69% eran varones (n=24). La edad en el momento de presentación era de 10±2 años (rango, 5–15). En la presentación clínica se incluían síntomas hepáticos en el 69% de los casos (n=24), hematológicos en el 11% (n=4) y neurológicos en el 3% (n=1). Seis (17%) eran hermanos asintomáticos. Seis niños (17%) fallecieron a causa de un fallo hepático fulminante. Se realizaron biopsias hepáticas en 21 pacientes (60%), que revelaron la presencia de cirrosis en 5 de ellos. Se llevó a cabo un trasplante hepático en 5 pacientes debido a una fallo hepático agudo.
ConclusionesLa EW debe sospecharse en niños con anormalidades persistentes de la función hepática. La presentación clínica de pacientes autóctonos fue similar a la descrita en poblaciones pediátricas de otros países.
Costa Rica is known to have a relatively high prevalence of Wilson's disease (WD). The present study describes the clinical presentation and demographic characteristics of patients diagnosed with WD at the Country's National Pediatric Hospital.
MethodsWe performed a retrospective chart review of all patients diagnosed with WD from January 1992 to March 2006.
ResultsThirty-five patients were diagnosed with WD and 69% were male (n=24). Age at presentation was 10±2 years (range, 5–15). Clinical presentation included hepatic features in 69% (n=24), hematological features in 11% (n=4) and neurological features in 3% (n=1). Six patients (17%) were asymptomatic siblings. Six children (17%) died from fulminant hepatic failure. Liver biopsies were performed in 21 patients (60%), showing cirrhosis in five. Five patients underwent liver transplantation due to acute liver failure.
ConclusionsWD should be suspected in children with chronically abnormal liver function tests. The clinical presentation of autochthonous patients was similar to that in children diagnosed in other countries.
La enfermedad de Wilson (EW) es un trastorno metabólico heredado en forma autosómica recesiva. El gen defectuoso se localiza en el brazo largo del cromosoma 131, y afecta al metabolismo del cobre, con una acumulación excesiva de este elemento en los riñones, el sistema nervioso central, los ojos y, especialmente, el hígado2. La incidencia general es de 1:30.000, proporción que en la población japonesa alcanza un valor de 1:10.000.
El establecimiento del diagnóstico no es siempre fácil, y representa un reto para los médicos debido al amplio espectro de la enfermedad y a la falta de métodos diagnósticos simples. Se han descrito numerosas presentaciones clínicas: enfermedad hepática, enfermedad psiquiátrica, anemia hemolítica, convulsiones, anormalidades neurológicas, disfunción renal y manifestaciones oculares. Múltiples parámetros bioquímicos e histológicos, como la excreción de cobre en orina de 24h en el momento de la presentación y tras la prueba de reto con D-penicilamina, así como la presencia de hallazgos típicos en las mitocondrias en el tejido hepático, podrían ayudar a establecer el diagnóstico3. El análisis genético provee un diagnóstico más definitivo y confirmatorio; sin embargo, no está disponible en todos los centros.
Costa Rica cuenta con un único centro de atención terciaria para la población pediátrica, que provee servicios a niños con enfermedades graves. Revisamos los expedientes del Hospital Nacional de Niños de Costa Rica para describir la presentación clínica y los resultados a largo plazo en niños diagnosticados de EW.
MétodosSe hizo una revisión retrospectiva de los expedientes de todos los pacientes diagnosticados de EW en el Hospital Nacional de Niños «Doctor Carlos Sáenz Herrera», en San José, Costa Rica, desde enero de 1992 hasta marzo de 2006. Se obtuvo la aprobación del comité de etica de la institución para la realización del estudio.
Las características demográficas recogidas incluyeron la edad de presentación, el sexo y el origen geográfico. En la presentación clínica se incluyeron, entre otras, las afecciones hepáticas, neurológicas y psiquiátricas. Se registró el manejo terapéutico y la aparición de complicaciones, así como la presencia de anillo de Kayser-Fleischer (K-F). Los parámetreos bioquímicos analizados incluyeron la concentración sérica de aspartato aminotransferasa (AST), alanino aminotrasferasa (ALT), bilirrubina conjudgada, albúmina sérica, hemoglobina y tiempo de protrombina. Asimismo, se registraron los valores de ceruloplasmina sérica y de la excreción de cobre en orina de 24h, en el momento basal y tras la prueba de reto con D-penicilamina (500mg v.o. inmediatamente antes y 12h después de la obtención de la muestra de orina). Se incluyeron los hallazgos histológicos en la biopsia hepática en caso de estar disponibles. El diagnóstico de EW se basó en la presencia de al menos dos o más resultados anormales en el metabolismo del cobre, junto con la evidencia clínica de la enfermedad y la respuesta a tratamiento quelante.
Se sumaron las variables categóricas expresadas como frecuencias y porcentajes, y las variables continuas expresadas como media±desviación estándar (DE) y, en caso de números sesgados, como media y rango. El test exacto de Fisher se utilizó para comparar las proporciones entre subgrupos de pacientes. El test de la t de Student se utilizó para comparar las medias de las variables continuas. Todos los tests fueron bilaterales, y se consideró estadísticamente significativo un valor de p<0,05. El análisis de los datos se realizó mediante el programa SPSS v. 15.0 (SPSS, Inc., Chicago, IL).
ResultadosTreinta y cinco niños fueron diagnosticados de EW desde enero de 1992 hasta marzo de 2006. Un 69% (n=24) eran varones (p=0,04), con una edad media de presentación de 10±2 años (rango, 5–15). La mayoría de los pacientes provenían de la ciudad de San José.
La presentación hepática se detectó en el 69% de los casos (n=24), e incluyó la elevación de los valores de ALT y AST, así como la presencia de hiperbilirrubinemia. Los parámetros bioquímicos revelaron una media de ALT de 167±355U/l (rango normal, 15–45), AST de 255±441U/l (rango normal, 10–40) y bilirrubina conjugada de 69±114μmol/l (rango normal, 0–3,4). Los valores de albúmina sérica se registraron en 28 niños, con una media de 29±9,3g/l (rango normal, 35–55).
El valor promedio de ceruloplasmina sérica fue de 23,8±42U/l (rango normal, 55–150). Los valores de excreción de cobre basal en orina de 24h estaban disponibles en 27 pacientes. Estos valores oscilaban entre 0,06 y 24±6μmol/24h (media, 6,1; valores normales<0,6). La prueba de reto con D-penicilamina de realizó en 10 pacientes, aunque sólo el 40% alcanzó el valor diagnóstico de 25μmol/24h. A 27 niños se les realizó una evaluación oftalmológica; el anillo de K-F se puso de manifiesto en un 22% de los casos (6 de 27). Solamente 4 pacientes (15%) tenían evidencia de anillo de K-F, ceruloplasmina baja y anormalidades bioquímicas consistentes con hepatitis.
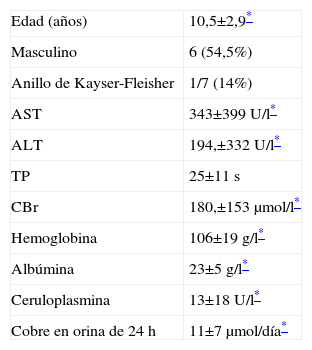
Once niños (31%) presentaron un fallo hepatico agudo, 5 de los cuales fueron trasplantados exitosamente y 6 fallecieron. Las características clínicas están descritas en la tabla 1 (apéndice).
Fallo hepático agudo. Características clínicas en 11 pacientes
| Edad (años) | 10,5±2,9* |
| Masculino | 6 (54,5%) |
| Anillo de Kayser-Fleisher | 1/7 (14%) |
| AST | 343±399U/l* |
| ALT | 194,±332U/l* |
| TP | 25±11s |
| CBr | 180,±153μmol/l* |
| Hemoglobina | 106±19g/l* |
| Albúmina | 23±5g/l* |
| Ceruloplasmina | 13±18U/l* |
| Cobre en orina de 24h | 11±7μmol/día* |
AST: aspartato aminotrasnferasa; ALT: alanino aminotrasnferasa; TP: tiempo de protrombina; CBr: bilirrubina conjugada.
Se constató una prolongación del tiempo de protrombina (TP) en el 90% de los pacientes, con una media de 25±5s (rango normal, 12–15). Los valores de la razón internacional normalizada (INR) estuvieron disponibles en 12 niños, sólo con una media de 2,82±2,7 (rango normal, 0,8–1,2).
Se observó una afección hematológica en 4 niños (11%), y 2 de ellos tuvieron anemia no hemolítica (hemoglobina<12g/l); todos ellos tenían trombocitopenia (recuento de plaquetas<150.000/l). Se apreció una ceruloplasmina baja y una elevación sutil de las enzimas hepáticas en 3 de ellos. Dos pacientes presentaron una excreción urinaria anormal de cobre. El tratamiento incluyó D-penicilamina y sales de cinc en 2 pacientes, uno recibió monoterapia con D-penicilamina y otro con sales de cinc.
Solamente un paciente presentó una afección neurológica a los 12 años de edad; los hallazgos incluyeron anemia, trobocitopenia y esplenomegalia. La historia clínica reveló una regresión de la función motora gruesa, un escaso desarrollo escolar y un progreso rápido hacia la espasticidad y disartria. La ultrasonografía abdominal mostró una disminución del tamaño del hígado. El valor sérico de ceruloplasmina fue de 0U/l y el examen oftalmológico reveló la presencia de anillo de K-F. Se realizó una biopsia hepática percutánea tras la corrección de la coagulopatía con vitamina K i.v. y una infusión de plasma fresco congelado, la cual confirmó el diagnóstico de EW. Se inició terapia quelante con dosis bajas de D-penicilamina y sales de cinc; sin embargo, la afección neurológica no ha cambiado de forma significativa tras 2 años de seguimiento.
Seis pacientes (17,1%) eran niños asintomáticos con una historia familiar de EW. La edad media fue de 9±2 años (rango, 6–15). Todos tuvieron valores bajos de ceruloplasmina sérica, con una media de 24±20U/l (rango, 0–56). Los valores de las transaminasas resultaron anormales en el 50% de los pacientes. Los valores séricos de albúmina y TP fueron normales en todos ellos. La excreción de cobre en orina de 24h estuvo elevada en todos los pacientes, con una media de 3±2μmol/l. La prueba de reto con D-penicilamina resultó negativa en los 3 niños en que se realizó. Ninguno presentó anillo de K-F. A 5 de los niños asintomáticos se les realizó una biopsia hepática, 4 presentaban esteatosis moderada y fibrosis, y uno tuvo cambios inespecíficos. El tratamiento incluyó el uso de D-penicilamina en 3 niños, y en los 3 restantes se asoció D-penicilamina con sales de cinc.
Las complicaciones se manifestaron en 5 niños con afección hepática y sangrado digestivo (14,4%); 6 niños fallecieron por un fallo hepático fulminante. Se hallaron diferencias estadísticamente significativas entre estos pacientes en la bilirrubina (10,6±9 frente a 1,5±3,1μmol/l; p<0,001) y la albúmina sérica (2,3±0,5 frente a 3,2±1,0g/l; p=0,02) en comparación con el resto del grupo.
Se realizaron biopsias hepáticas en 21 pacientes (60%), que revelaron cambios histológicos graves en 8 (esteatosis extensa y/o inflamación o necrosis que afectaba a la mayoría del parénquima y/o fibrosis extensa), así como cirrosis en 5 (23,8%). Casi un 33% de los registros histológicos revelaron hallazgos leves-moderados (esteatosis leve, inflamación y fibrosis leve). La presencia de hallazgos histológicos graves se asoció a la aparición de complicaciones, como sangrado, muerte y necesidad de trasplante (p=0,04). Ninguno de los registsros de biopsias estableció la cuantificación de cobre en tejido hepático.
La mayoría de los pacientes (n=32) recibió terapia quelante con D-penicilamina (15–20mg/kg/día, 2–3 veces al día), y en 14 se combinó con sales de cinc. Solamente un paciente recibió monoterapia con sales de cinc. Nueve niños con fallo hepático fulminante recibieron inicialmente D-penicilamina y 5 fueron trasplantados eventualmente.
DiscusiónLa incidencia estimada de EW es de 1: 30.0004. En Costa Rica la prevalencia estimada en la población general es de 6:100.0005. En el único hospital pediátrico de Costa Rica, 35 niños fueron diagnosticados en un período de 14 años, incluidos 6 hermanos asintomáticos.
Hasta ahora se han identificado más de 200 mutaciones de ATP7B, el gen responsable del desarrollo de EW, pero solamente unas pocas son más frecuentes en grupos poblacionales específicos. Un estudio previo realizado en Costa Rica identificó la secuencia del defecto, lo que llevó al reconocimiento de la presencia de la misma mutación en una población de Sardinia, Italia5,6. Hay registros históricos de la colonización de Costa Rica hace siglos por parte de individuos de Sardinia, y el análisis genealógico demostró que dichos individuos trajeron el gen mutante a Centroamérica. La alta incidencia podría explicarse debido a que hay poco movimiento poblacional en Costa Rica.
La EW representa un reto para los médicos. Cualquier niño que presente síntomas de hepatitis, así como una elevación de las transaminasas, debería ser evaluado. En nuestro estudio hubo 11 niños que presentaron un fallo hepático fulminante (31%), a 5 de los cuales se les realizó un trasplante hepático de forma exitosa. Estos datos son similares a los indicados en el estudio de Dhawan et al7, en el que el fallo hepático fulminante afectó al 33% de la población pediátrica con EW. La tríada de ceruloplasmina baja, anillo de K-F y hepatitis aparece en algunos pacientes en edad pediátrica. Algunos autores han reportado que el 30–50% de los casos presentan estas características2,8, lo que contrasta con nuestro estudio, en el que solamente el 15% de los pacientes presentó dicha tríada. Esto podría estar relacionado con la edad temprana en el momento del diagnóstico, así como con falsos valores normales de ceruloplasmina.
La ceruloplasmina usualmente está reducida en la mayoría de pacientes con EW. Sin embargo, tiene un valor predictivo positivo bajo, debido a que sólo el 20% de estos pacientes presenta valores bajos de ceruloplasmina sérica. Un 68% de nuestra población presentó valores de ceruloplasmina<20 mg/dl; sin embargo, hasta un 90% presentó cifras por debajo del valor normal de referencia, por lo cual continúa siendo de utilidad para orientar el diagnóstico. De los 6 hermanos asintomáticos, sólo 2 tuvieron valores bajos de ceruloplasmina sérica. Se sabe que los valores de ceruloplasmina pueden ser bajos, normales o altos en pacientes con EW. Ciertas condiciones especiales, como el fallo hepático, el embarazo y la corta edad9 pueden representar un mayor problema para el ejercicio diagnóstico.
La medición de la excreción de cobre en urina de 24h es una herramienta de valor en el diagnóstico de EW; en nuestro estudio el 85% de los pacientes (23 de 27) la presentaban cifras elevadas. Esto es comparable con lo reportado previamente en otro estudio, en el que se indicó que la mayoría de los pacientes con EW sintomática tenían unos valores elevados de cobre basal4.
La presencia de anillos de K-F y ceruloplasmina baja son suficientes para diagnosticar EW1,10,11. En un estudio pediátrico, el anillo de K-F se observó en el 100% de los apcientes; sin embargo, estos niños presentaron un fallo hepático fulminante12. En el presente estudio se constató la presencia de anillo de K-F en 6 de 27 niños (22%), incluido el caso con afección neurológica. Este hallazgo es similar al reportado en un estudio retrospectivo realizado en España, en el que sólo 3 niños (19%), entre 26 casos, presentaron anillo de K-F13.
En nuestro estudio un 11,4% de los pacientes presentó una afección hematológica, que incluía trombocitopenia y anemia no hemolítica, porcentaje bajo comparado con el de otros ensayos. Stremmel et al14 obtuvieron hallazgos hematológicos en el 31% de los pacientes, en quienes la anormalidad más común fue la trombocitopenia; esto podría explicarse por la presencia de una enfermedad más avanzada e hiperesplenismo basado en hallazgos de biopsia hepática; dos tercios de estos pacientes tenían fibrosis o cirrosis hepática.
Los síntomas neurosiquiátricos son raros en la edad pediátrica; sin embargo, algunos autores indican que hasta un 60% de los pacientes evaluados presentaron disartria, tremor y dificultades para la escritura, entre otros síntomas14, en contraste con nuestra serie de casos, en la que solamente un paciente tuvo una manifestación neurológica. Los síntomas leves, como cefalea, nerviosismo y falta de atención, no se anotan detalladamente en las historias clínicas, lo que podría explicar estas diferencias.
Las biopsias hepáticas son útiles, aunque no se realizan habitualmente en los niños debido a que implica un riesgo de sangrado en un paciente que podría tener una coagulopatía; en dicho caso, estaría indicada la biopsia transyugular de no corregirse el defecto en la coagulación. En nuestra serie de casos hubo una asociación estadísticamente significativa entre los hallazgos histológicos moderados o graves con la aparición de complicaciones, como muerte o necesidad de trasplante hepático. Desafortunadamente, no se determinó la concentración de cobre en el tejido hepático en los pacientes a quienes se les realizó una biopsia, debido a que no se contaba con recursos locales para dicha medición, lo cual hubiera sido de gran utilidad diagnóstica.
La iniciación de tratamiento quelante es crucial en estos pacientes debido a que la progression de la enfermedad conlleva resultados fatales. La D-penicilamina fue el único agente quelante disponible en nuestro centro; las sales de cinc se utilizaron como terapia adyuvante. Los efectos secundarios, como hipersensibilidad dérmica, proteinuria, supresión de médula ósea y enfermedades autoinmunes, deben tenerse en cuenta cuando se utiliza D-penicilamina15.
El trasplante hepático se ha convertido en otra modalidad terapéutica y ha salvado la vida de muchos pacientes con fallo hepático fulminante16. El presente estudio ninguno de los pacientes con otro tipo de presentación clínica y en terapia quelante crónica requirió trasplante hepático, lo que contrasta con lo descrito por otros autores8,17.
El curso clínico depende de la gravedad de la enfermedad en el momento del diagnóstico; sin embargo, la recuperación completa es factible una vez se inicia la terapia quelante. Nuestra experiencia es similar a la reportada en Estados Unidos y Europa respecto a la EW pediátrica2, aunque creemos que los estudios genéticos ayudarán a correlacionar las diversas presentaciones fenotípicas.
En resumen, el diagnóstico de EW en poblaciones pediátricas es un reto; sin embargo, la sospecha clínica es clave en la búsqueda de un diagnóstico certero y para establecer un tratamiento eventual. Es necesario realizar un cribado de hermanos asintomáticos debido a que podrían presentar cambios histológicos graves con desenlaces fatales. Las complicaciones graves se relacionan con la presencia de hipoalbuminemia, la elevación de la bilirrubina conjugada y la cirrosis hepática. Costa Rica es un país pequeño, donde se ha descrito previamente una alta incidencia de EW; sin embargo, la presentación clínica de esta enfermedad es similar a la reportada en otros estudios.
Los autores agradecen al Dr. David Mack la revision de este manuscrito, y a Nick Barrowman, PhD, la realización de los cálculos estadísticos.

