



La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa que se ha descrito tradicionalmente con expresión clínica de afección a la neurona motora superior en el homúnculo motor y a la neurona motora inferior en la médula espinal1,2. Sin embargo, recientemente se han descrito en sujetos con ELA definida, signos y síntomas extra-motores; incluyendo deterioro en las funciones cerebrales superiores, signos disautonómicos, trastornos metabólicos3,4, así como alteraciones cognitivas asociadas a la demencia frontotemporal5. Estudios histopatológicos en tejidos de autopsia de sujetos con ELA definida-esporádica, han identificado inclusiones proteínicas de transactive response DNA binding protein 43 kDa (TDP-43) en áreas no motoras del sistema nervioso central, incluyendo el sistema nigroestriatal, cerebelo, prosencéfalo, hipotálamo, así como áreas neocorticales y alocorticales6,7. La ELA al igual que la demencia frontotemporal son actualmente consideradas proteinopatías TDP-43.
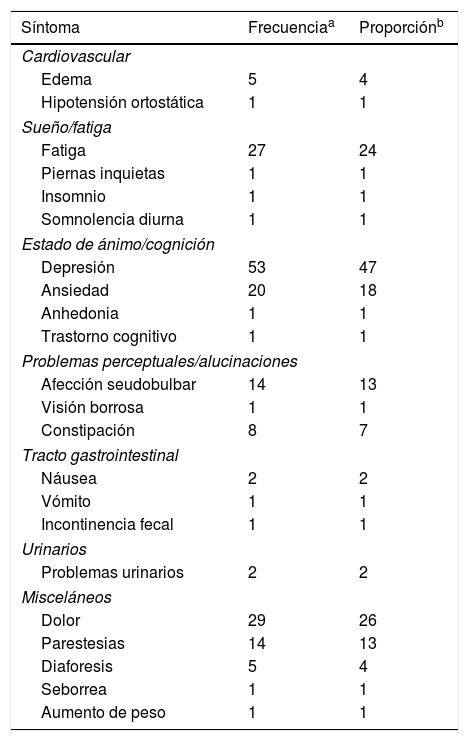
Realizamos un análisis retrospectivo en expedientes clínicos de 112 pacientes con el diagnóstico establecido de ELA definida-esporádica, de acuerdo a los criterios clínicos y neurofisiológicos de El Escorial8. Se analizaron los signos y síntomas neurológicos extra-motores reportados al momento del diagnóstico, al igual que las manifestaciones extramotoras que se presentaron dentro del primer año de seguimiento clínico. En todos los pacientes con diagnóstico de ELA definida se realizó, además de la escala funcional estratificada revisada para ELA (ALSFRS-r), examen mini-mental, test neuropsicológico, test genético y escala de Hamilton para ansiedad y depresión en la consulta inicial, y al menos una evaluación adicional en el año de seguimiento. En nuestra cohorte de pacientes se identificaron 25 síntomas extra-motores, los más prevalentes fueron; depresión (47%), dolor (26%), fatiga (24%), ansiedad (18%), afección seudobulbar (13%) y parestesias (13%) (tabla 1). No se estableció el diagnóstico de demencia frontotemporal en ninguno de nuestros pacientes y curiosamente todos los casos con afección seudobulbar, depresión o ansiedad resultaron negativos para la expansión del hexanucleótido del cromosoma 9 (gen C9Orf72), habitualmente asociado a sintomatología seudobulbar. Encontramos un promedio de 4,2±2,03 síntomas extra-motores por paciente. Los pacientes con ELA bulbar presentaron mayor número de síntomas extra-motores comparados con sujetos con ELA espinal, 5,41±1,61 y 3,85±2,01 síntomas extra-motores por paciente, respectivamente. Adicionalmente, se observó una correlación como variables dependientes entre pares de síntomas que podrían tener un mecanismo fisiopatológico similar incluyendo; ansiedad y depresión (χ2; p=0,025), incontinencia urinaria y constipación (χ2; p=0,017), así como dolor y parestesias (χ2; p=0,027).
Síntomas extra motores en ELA clasificados por dominio, frecuencia y valor proporcional
| Síntoma | Frecuenciaa | Proporciónb |
|---|---|---|
| Cardiovascular | ||
| Edema | 5 | 4 |
| Hipotensión ortostática | 1 | 1 |
| Sueño/fatiga | ||
| Fatiga | 27 | 24 |
| Piernas inquietas | 1 | 1 |
| Insomnio | 1 | 1 |
| Somnolencia diurna | 1 | 1 |
| Estado de ánimo/cognición | ||
| Depresión | 53 | 47 |
| Ansiedad | 20 | 18 |
| Anhedonia | 1 | 1 |
| Trastorno cognitivo | 1 | 1 |
| Problemas perceptuales/alucinaciones | ||
| Afección seudobulbar | 14 | 13 |
| Visión borrosa | 1 | 1 |
| Constipación | 8 | 7 |
| Tracto gastrointestinal | ||
| Náusea | 2 | 2 |
| Vómito | 1 | 1 |
| Incontinencia fecal | 1 | 1 |
| Urinarios | ||
| Problemas urinarios | 2 | 2 |
| Misceláneos | ||
| Dolor | 29 | 26 |
| Parestesias | 14 | 13 |
| Diaforesis | 5 | 4 |
| Seborrea | 1 | 1 |
| Aumento de peso | 1 | 1 |
En un estudio reciente, Cykowski et al. demostraron inclusiones de TDP-43 en el prosencéfalo e hipotálamo, reportando una correlación significativa de este hallazgo histopatológico en sujetos con ELA de predominio bulbar7. En nuestra cohorte los pacientes con predominio bulbar presentaron mayor número de síntomas extra-motores al compararse con los pacientes de ELA espinal.
La ELA es una enfermedad de presentación clínica muy heterogénea9; la presencia de inclusiones patológicas en áreas extra-motores del sistema nervioso central observadas con ELA definida-esporádica podrían explicar la presencia de síntomas atípicos, frecuentemente omitidos y que por consecuencia no se abordan ni se tratan de forma adecuada, limitando la calidad de vida del paciente. En la presente cohorte, los síntomas extra-motores observados en los pacientes con ELA, podrían ser manifestaciones incidentales, consecuencia de la discapacidad o expresiones clínicas por progresión del proceso neurodegenerativo. La identificación oportuna de manifestaciones extramotoras en pacientes con los síntomas motores clásicos, podría facilitar su tratamiento médico y mejorar la condición clínica de los pacientes con ELA. La variabilidad de los fenotipos clínicos, los recientes hallazgos histopatológicos extra-motores en el SNC y la presencia de síntomas no-motores, sugieren que la ELA es una enfermedad multisistémica no restringida al sistema motor.

