


La asociación entre factores ambientales presentes durante el desarrollo embrionario/fetal y enfermedades que puedan presentarse durante la vida representa un campo de creciente interés. En este contexto la evidencia actual apoya fuertemente que alteraciones en el crecimiento intrauterino y durante los primeros años de vida presentan una fuerte influencia en el riesgo de padecer enfermedades crónicas que en muchos casos pudiera ser mayor que la carga genética del paciente. La persistencia y reproducibilidad de los fenotipos asociados a alteraciones en el desarrollo temprano sugieren la participación de mecanismos moleculares que registran dichas modificaciones (i.e. mecanismos epigenéticos) generando una «reprogramación» celular y fisiológica. Esta revisión es la introducción a una serie de 5 artículos en torno a la participación de los mecanismos epigenéticos en el desarrollo de enfermedades crónicas (i.e. cardiovasculares, metabólicas, asma/alergias y cáncer) y su relación con el origen de dichas enfermedades en etapas tempranas del desarrollo. El objetivo de esta serie es mostrar el estado actual de esta área de la investigación y presentar los desafíos e interrogantes futuros en los cuales la pediatría tiene un papel preponderante, desarrollando estrategias para la prevención, detección precoz y seguimiento.
Current evidence supports the notion that alterations in intrauterine growth and during the first years of life have a substantial effect on the risk for the development of chronic disease, which in some cases is even higher than those due to genetic factors. The persistence and reproducibility of the phenotypes associated with altered early development suggest the participation of mechanisms that would record environmental cues, generating a cellular reprogramming (i.e. epigenetic mechanisms). This review is an introduction to a series of five articles focused on the participation of epigenetic mechanisms in the development of highly prevalent chronic diseases (i.e. cardiovascular, metabolic, asthma/allergies and cancer) and their origins in the foetal and neonatal period. This series of articles aims to show the state of the art in this research area and present the upcoming clues and challenges, in which paediatricians have a prominent role, developing strategies for the prevention, early detection and follow-up.
En su obra El origen de las especies mediante la selección natural Darwin estableció las bases para la comprensión de la existencia y éxito de las diversas especies. A pesar de la mirada estrictamente genética que derivó de su propuesta, y que moldeó la ciencia biomédica durante el siglo xx, Darwin propone en el sexto capítulo que la «ley superior» del origen de las especies sería la adaptación a «las condiciones de existencia» más que la «selección natural» de características heredables (i.e. genes)1. En otras palabras, la capacidad de interaccionar y responder adecuadamente a las fluctuaciones constantes del ambiente condiciona en mayor medida las posibilidades de un individuo de transmitir sus genes más que las propiedades intrínsecas de los genes2. En este contexto Conrad Waddington, en sus estudios de embriología, infirió tempranamente la importancia de la interacción gen-ambiente, a través de la cual a partir de un genoma único se generan los distintos tipos y funciones celulares que constituyen un organismo, mediante estímulos que se suceden en un tiempo y lugar específico3. Esta capacidad del medioambiente de moldear el fenotipo fue inicialmente definida por Waddington como «epigenética»4.
La definición del término «epigenética» resulta conflictiva, de hecho actualmente existen 2 usos de dicho concepto5: 1) para explicar cambios en la herencia de fenotipos que no son atribuibles a cambios en la secuencia del ADN; y 2) para explicar cómo un organismo genera diversos fenotipos celulares a partir de un único genotipo. Tomando en cuenta los mecanismos considerados como epigenéticos, ambas definiciones no son integrativas. Sin embargo, en los últimos años se ha propuesto una definición más amplia de epigenética5,6, en la que se considera como «mecanismos modificadores de cromosomas que cambian la plasticidad fenotípica de una célula u organismo». A pesar de las diferentes definiciones se encuentra bien establecido que los mecanismos epigenéticos tienen una función clave en el desarrollo, no solo controlando la diferenciación celular, sino además registrando señales del medioambiente en condiciones fisiológicas4, patológicas7 y durante el envejecimiento8. Así mismo creciente evidencia muestra cómo modificaciones en los patrones epigenéticos durante los primeros años de vida condicionan el riesgo de desarrollar enfermedades crónicas no heredables.
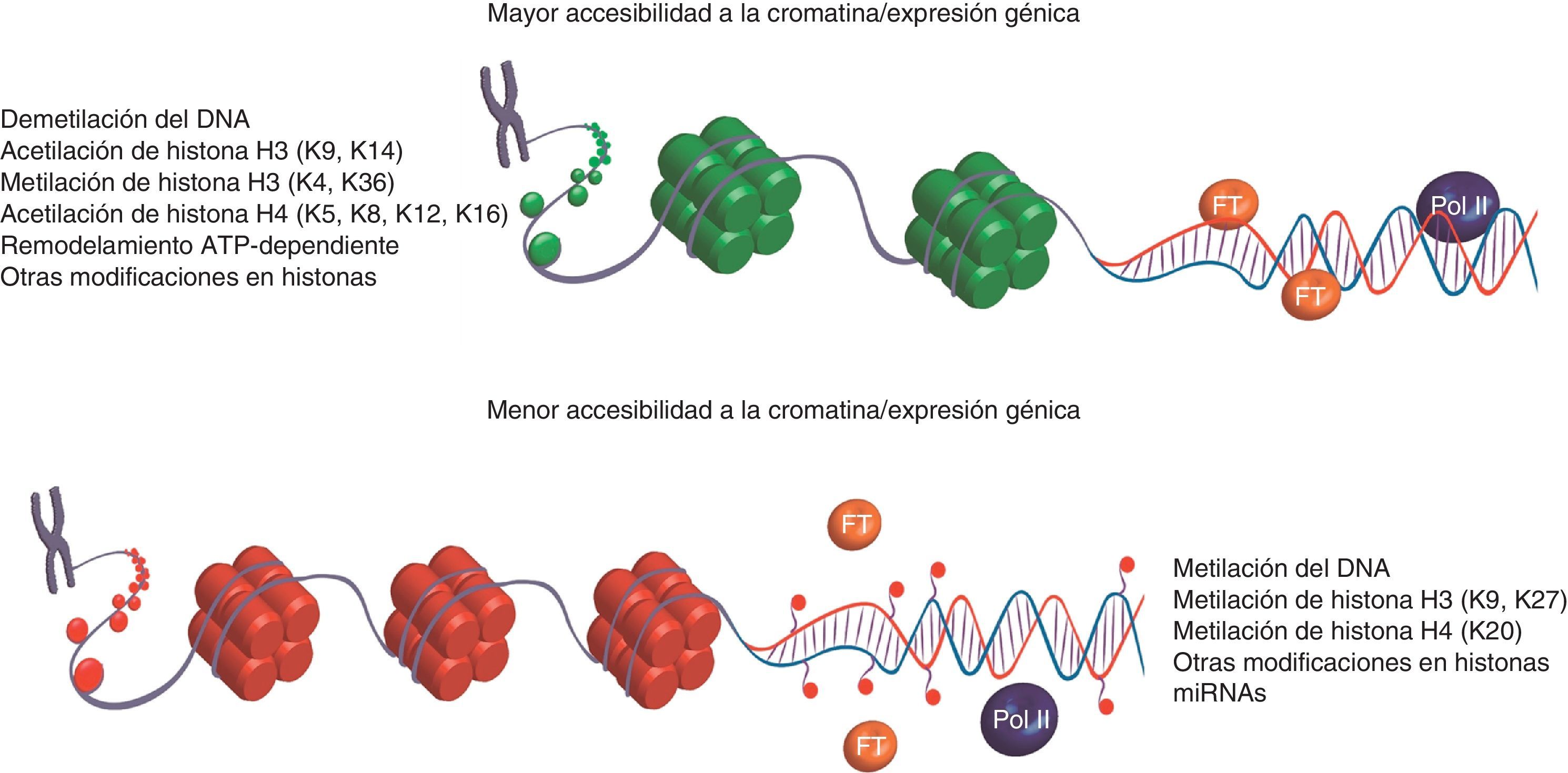
Mecanismos epigenéticosEn las células eucariontes el ADN se encuentra confinado en el núcleo y plegado parcialmente en un complejo proteína-ADN denominado «nucleosoma»9. Este nucleosoma está constituido por un octámero de proteínas llamadas histonas, cubiertas por 146pb de ADN. Dicha estructura puede ser transformada por modificaciones tanto a nivel del ADN como de las histonas, generando regiones del ADN expuestas en mayor o menor grado a la maquinaria transcripcional. Los fenómenos moleculares que cambian estas estructuras, ADN y/o histonas, son denominados modificaciones epigenéticas. Los múltiples tipos de modificaciones epigenéticas existentes conducen a un nivel complejo y dinámico de interacciones y estructuras proteína-ADN que permiten la regulación de la expresión de genes a corto y a largo plazo10. Los mecanismos moleculares considerados epigenéticos incluyen metilación del ADN, modificaciones postraduccionales de histonas, modificación de cromatina dependiente de ATP y ARN no codificantes (fig. 1).
 y la polimerasa de ARN (Pol II) sin mutar la secuencia primaria del ADN, así como la unión del ADN a las histonas (octámeros verdes y rojos). En la parte superior se indican aquellas modificaciones epigenéticas asociadas positivamente con la accesibilidad a la cromatina (histonas en verde), permitiendo la unión de la maquinaria transcripcional y la expresión génica. En contraste los mecanismos epigenéticos enlistados en la parte inferior reducen la accesibilidad a la cromatina (histonas en rojo), generando regiones con una estructura «cerrada» o evitando la unión de factores de transcripción al ADN mediante su metilación (círculos rojos unidos mediante una cinta púrpura al ADN).")
Mecanismos epigenéticos y sus efectos sobre la expresión de genes. La expresión de genes puede ser controlada a mediano y largo plazo modificando tanto los sitios de unión de factores de transcripción (FT) y la polimerasa de ARN (Pol II) sin mutar la secuencia primaria del ADN, así como la unión del ADN a las histonas (octámeros verdes y rojos). En la parte superior se indican aquellas modificaciones epigenéticas asociadas positivamente con la accesibilidad a la cromatina (histonas en verde), permitiendo la unión de la maquinaria transcripcional y la expresión génica. En contraste los mecanismos epigenéticos enlistados en la parte inferior reducen la accesibilidad a la cromatina (histonas en rojo), generando regiones con una estructura «cerrada» o evitando la unión de factores de transcripción al ADN mediante su metilación (círculos rojos unidos mediante una cinta púrpura al ADN).
En mamíferos el ADN es metilado principalmente en residuos de citosina presentes en dinucleótidos citosina-guanosina (CpG), a través de una reacción que transfiere un grupo metilo desde el dador S-adenosil-L-metionina a la posición 5′ del anillo de citosina, generando 5-metilcitosina11. Esta reacción es catalizada por un grupo de enzimas denominadas metiltransferasas de ADN (DNMT) compuestas por 2 familias DNMT1 y DNMT3 (3a y 3b), codificadas por 3 genes distintos11. La DNMT1 mantiene el patrón de metilación del ADN durante la división celular mitótica y después de la fertilización12. Este proceso es guiado por la presencia de CpG hemi-metilados, los cuales son identificados por la DNMT1 en el ADN de doble hebra13. La actividad de esta enzima explica por qué la metilación del ADN ha sido considerada una modificación epigenética altamente heredable. Sin embargo, la DNMT1 presenta una baja eficiencia en mantener el patrón de metilación en regiones que presenta una elevada presencia de CpG (islas CpG)14, y durante algunas etapas del desarrollo (i.e. después de la fertilización) ocurre un proceso de demetilación masiva12, lo cual puede generar modificaciones en el patrón de metilación. Las otras 2 metiltransferasas, DNMT3a y DNMT3b, catalizan la metilación de novo que permite la generación de nuevos patrones de metilación durante la gametogénesis, el desarrollo embrionario y la diferenciación celular15,16. Ambas enzimas poseen propiedades cinéticas similares11,13; sin embargo, sus actividades participan en diferentes procesos. La DNMT3a tiene una función primordial en generar patrones de metilación durante la gametogénesis en los denominados «genes improntables» (imprinted genes), mientras la DNMT3b es importante durante el desarrollo embrionario. En casi todos los casos la metilación del ADN representa un marcador de silenciamiento de genes a largo plazo17,18. Este silenciamiento ocurre principalmente a través de: 1) una reducción en la unión de factores de transcripción cuando la modificación se encuentra en un sitio de unión para el factor en específico; o 2) el reclutamiento de represores como proteínas de unión a CpG (MCP) y deacetilasas de histona (HDAC)17.
Modificaciones postraduccionales de histonasLa proteína estructural de los nucleosomas está constituida por 2 copias de 4 proteínas denominadas histona 2A (H2A), H2B, H3 y H4, las cuales presentan una estructura similar que posibilita su interacción con el ADN9. Adicionalmente, estas proteínas presentan un dominio globular mediante el cual interactúan para formar el octámero, y una cola flexible que participa activamente en la interacción con el ADN19. A diferencia de la metilación del ADN, las modificaciones en las histonas son más dinámicas y no se relacionan directamente con un efecto definido (activación o represión). Las modificaciones de histonas se relacionan con el contexto en el que ellas se encuentran, o dicho de otra manera, la presencia de otras modificaciones de histonas, sugiriendo la existencia de un «código de histonas»6,10,19, el cual determinaría el efecto activador o represor sobre la expresión de un gen. Hasta la fecha han sido identificadas más de 50 enzimas que catalizan diversas modificaciones de histonas20,21, las cuales han sido clasificadas de acuerdo al tipo de reacción que llevan a cabo. Las modificaciones de histonas mejor comprendidas son aquellas relacionadas con la acetilación/deacetilación y metilación/demetilación de histonas.
La acetilación de histonas ocurre en residuos de lisina (K) e involucra la transferencia de un grupo acetilo desde la acetil-CoA. En mamíferos esta reacción es llevada a cabo por 3 familias de acetil-transferasas de histonas (HAT), denominadas GNAT, MYST y CBP/p30021. Esta modificación se considera un activador de la expresión que estabiliza la carga básica de la lisina en la histona disminuyendo su afinidad por el ADN, lo cual evita la formación de una cromatina altamente compactada21,22. Las acetilaciones de histonas mejor caracterizadas son aquellas que ocurren en los residuos de lisina 9 (K9), K14, K18 y K56 en H3, y K5, K8, K13 y K16 en H418. Por lo menos 4 clases de deacetilasas de histonas (HDAC I, II, III y IV) han sido identificadas, las cuales catalizan las reacción reversa de las HAT21. Esta actividad enzimática se relaciona con el silenciamiento de genes, progresión del ciclo celular, diferenciación y la respuesta inducida por daño al ADN22. La actividad de las HDAC puede ser inducida en respuesta a metilación del ADN al reclutar a las MCP, las cuales poseen un sitio de interacción con varias HDAC, sugiriendo que el silenciamiento de genes puede resultar de una acción concertada entre modificaciones en el ADN e histonas17.
Ciertos residuos de lisina en H3 y H4 pueden ser modificados adicionalmente por metilaciones. Sin embargo, contrario a lo que ocurre en la metilación del ADN, el efecto de esta modificación no está relacionado con un resultado definido, sino más bien depende del residuo en específico donde ocurre. Además, un mismo residuo puede ser mono, di o trimetilado, y dependiendo del grado de metilación se puede generar una represión o activación del gen12. La H3 puede ser metilada en los residuos de K4, K9, K27, K36 y K79. Las modificaciones en las lisinas 4, 36 y 79 se encuentran asociadas con activación transcripcional, mientras aquellas presentes en K9 y K27 se relacionan con represión. Por otra parte, H4 presenta metilaciones solo en K20, la cual está asociada a una activación en la expresión18. Las metilaciones en histonas son removidas por demetilasas de histonas (HDMT); sin embargo, hasta la fecha se han identificado solo aquellas enzimas que presentan actividad sobre H323.
Modificación de cromatina dependiente de adenosín trifosfatoLa cromatina puede ser modificada en un proceso dependiente de ATP, en el cual la interacción ADN/histonas es liberada parcialmente (looping), permitiendo que la histona se deslice sobre el ADN (sliding), ubicándose en una nueva posición respecto de la cadena de ADN24. Este proceso posibilita además el reemplazo entre variantes de H2 modificadas y no modificadas. Este reemplazo puede desestabilizar la interacción con H4 llevando a una apertura de la cromatina19. Esta actividad se ha encontrado en 4 familias de proteínas (SWI/SNF, ISWI, NURD/Mi-2/CHD e INO80), las cuales se relacionan con replicación del ADN, progresión del ciclo celular, supresión de tumores, diferenciación, regulación del cromosoma X, entre otros fenómenos25.
Ácidos ribonucleicos no codificantesLa idea de que ARN no codificantes pudiesen regular la expresión de genes fue propuesta inicialmente en los años 196026; sin embargo, los mecanismos implicados en este fenómeno han comenzado a conocerse en los últimos años, entregando una función más dinámica al ARN, aparte de sus funciones clásicas en la transcripción y traducción. De hecho, menos de un 5% de los ARN que son transcritos codifican para alguna proteína, mientras la gran mayoría corresponde a ARN no-codificantes (ncARN), dentro de los cuales un gran porcentaje participa en mecanismos de regulación de expresión (ncARN reguladores)27,28.
Dentro de los principales ncARN reguladores se encuentran los ncARN «largos» (lncARN), pequeños ARN interferentes (siARN) y los micro ARN (miARN). Los lncARN regulan la expresión de un gen complementario específico, ya sea a través del remodelamiento de la cromatina, el procesamiento alternativo de los mARN (splicing) o la generación de siARN27. Estos últimos, junto a los miARN representan mecanismos epigenéticos basados en ARN de interferencia, que silencian genes a través de ARN no codificantes de ∼21pb. Hasta la fecha se han reportado más de 1.000 de estos «micro ARN» (miRAN), los cuales son transcritos por la polimerasa de ARN tipo ii y se encuentran en su mayoría (∼70%) codificados en genes específicos para ellos y, en menor cantidad, en las regiones intrónicas de genes que codifican para proteínas. Los miARN son transcritos como pre-miARN, siendo procesados en primera instancia en el núcleo por el complejo DROSHA-DGCR8. Posteriormente se exportan al citoplasma para su maduración a miARN mediante la acción del complejo formado por la proteína DICER1 y las ARNsa IIIa IIIb29.
De este procesamiento resulta un ARN de hebra simple, el cual es incorporado al «complejo proteico de silenciamiento inducido por miARN» (miRISC), el que se une a ARN mensajeros que presenten alguna región complementaria en su secuencia al miARN maduro. Se postula que una complementariedad total entre el miARN y el mARN conduce a su degradación del mARN, mientras que la complementariedad parcial reprime la traducción proteica30. Adicionalmente ciertos miARN participan en la eliminación de secuencias de ADN y silenciamiento de transposones31. De manera notable un único miARN puede regular la expresión de múltiples mARN, muchas veces asociados a vías de señalización o procesos metabólicos. Así mismo, varios miARN pueden confluir en la regulación de un mARN, constituyendo un mecanismo de regulación de la expresión génica complejo29,30.
En general, se ha reportado que los ncARN reguladores participan activamente en control del locus de Igf2r (grupo de genes que controla en gran parte el crecimiento fetal) y la inactivación del cromosoma X31. Adicionalmente, se ha reportado que ciertos ARN no codificantes complementarios al ADN inducen la metilación de regiones promotoras32. Por otra parte, debido a su estabilidad y a la posibilidad de ser detectados y cuantificados mediante PCR cuantitativa, la caracterización de miARN en diversos tipos de muestras se ha propuesto como una metodología diagnóstica. En este contexto nuestro grupo ha reportado recientemente la asociación entre el riesgo de desarrollar síndrome metabólico en niños de 10 a 12 años y los niveles en sangre de 2 miARN que regulan la vía de señalización de insulina, presentando a su vez estos niños mayores niveles de insulinemia en ayuno33.
El desarrollo temprano como condicionante a largo plazo de la salud: programación fetal y perinatalLa asociación entre factores ambientales presentes durante el desarrollo embrionario/fetal y enfermedades que puedan presentarse durante la vida representa un campo de creciente interés. Durante la última parte de los años 1980 estudios epidemiológicos en Gran Bretaña correlacionaron antecedentes clínicos perinatales (bajo peso de nacimiento o restricción de crecimiento intrauterino [RCIU] y rápida ganancia de peso infantil) con el desarrollo de enfermedades cardíacas34, intolerancia a la glucosa35, diabetes tipo 2 e hipertensión36. En este contexto la evidencia actual apoya fuertemente que alteraciones en el crecimiento intrauterino presentan una mayor influencia en el riesgo de desarrollo de enfermedades cardiovasculares a largo plazo37 que los antecedentes genéticos del paciente38. La persistencia y reproducibilidad de los fenotipos asociados a alteraciones en el desarrollo temprano sugiere la participación de mecanismos moleculares que registran dichas modificaciones (i.e. mecanismos epigenéticos) generando una «reprogramación» celular y fisiológica39,40. Dicho de otra manera, el concepto «programación fetal y perinatal» se puede considerar como el establecimiento de una respuesta modificada a nivel sistémico y celular, como respuesta a estímulos ambientales que tienen lugar en un momento determinado del desarrollo embrionario, fetal y neonatal41. Estos estímulos inducen cambios funcionales, los cuales suponen una adaptación a fin de «enfrentar de la mejor forma» las posibles condiciones ambientales existentes al nacer40,42,43.
Durante el periodo periconcepcional y preimplantacional factores como el estado nutricional y estrés de la madre, o niveles de hormonas y oxígeno, afectan el desarrollo del oocito y el blastocisto alterando el número de células destinadas a trofoblasto y el potencial de crecimiento de la placenta44,45. Se ha visto un menor potencial de crecimiento e implantación del blastocisto en madres diabéticas46, lo cual se observa al exponer in vitro embriones de ratón a elevadas concentraciones de D-glucosa (28mM)47,48. Por otra parte, la suplementación de la dieta materna con aminoácidos esenciales y no esenciales potencia el desarrollo placentario y el crecimiento fetal. Este último efecto podría estar asociado a una mayor expresión de moléculas de adhesión por parte del blastocisto, lo cual ha sido descrito in vitro49,50. Por otra parte, la placenta tiene una función crucial durante el periodo fetal. La placenta recibe e interpreta señales del feto y la madre generando respuestas que regulan principalmente la disponibilidad de nutrientes y factores de crecimiento para el feto51,52. En embarazos normales la placenta tiene una superficie total para intercambio de 11-12m2 al término de la gestación. Sin embargo, las características estructurales y funcionales de la placenta pueden verse modificadas de manera permanente por la exposición a determinados estímulos en periodos específicos del desarrollo53.
Evidencia de programación epigenéticaLa evidencia actual muestra que los mecanismos epigenéticos participan activamente durante el desarrollo temprano, tanto en condiciones normales como subóptimas. De hecho, la estructura y función de la placenta está determinada en gran parte por la expresión de los llamados imprinted genes (igf2, igf2r y peg1), cuya expresión es regulada por metilaciones del ADN de un modo «dependiente del progenitor»54,55. Por otra parte, la invasión de trofoblasto es limitada por la expresión de la proteína Maspin, cuya expresión es regulada dinámicamente por el intercambio de modificaciones postraduccionales en las histonas de su promotor56. Así mismo, acetilaciones en las histonas regulan la expresión de la hormona similar a la somatomamotrofina57, la cual se asocia a mayor crecimiento placentario y fetal. En enfermedades del embarazo, como preeclampsia y RCIU, se ha encontrado un incremento en la expresión de inhibidores de serín-proteasas inducidas por estrés (Serpin). Esto se ha asociado a hipometilación del ADN en los sitios de unión para el factor inducido por hipoxia (HIF) ubicados en el promotor del gen58. Adicionalmente, estudios en modelos animales demuestran que la interacción posnatal del recién nacido con el ambiente y con su madre activan mecanismos epigenéticos que programan tanto el metabolismo como la conducta, modificando la metilación del ADN de genes asociados a la respuesta al estrés59.
Por otra parte, se ha podido establecer la presencia de marcadores epigenéticos asociados con el riesgo de enfermedades crónicas y las condiciones de desarrollo temprano. Existe contundente evidencia de que tanto el crecimiento fetal deficiente (RCIU) como la macrosomia fetal se asocian a mayor riesgo de desarrollo de enfermedades crónicas. En este contexto, un estudio comparó la metilación global de ADN al nacer en células progenitoras hematopoiéticas de individuos controles, RCIU y grandes para la edad gestacional (GEG). De manera destacable se evidenció cambios considerables en los niveles de metilación a nivel genómico, tanto en células derivadas de RCIU como GEG comparados con los controles, presentándose una gran coincidencia en los promotores de genes afectados por ambas condiciones, pero una distinta localización en los CpG específicos afectados en cada caso60. Así mismo, un estudio pionero analizó la metilación del ADN de los promotores del factor de crecimiento similar a la insulina tipo 2, receptor de insulina y la enzima oxidante de ácidos grasos palmitoil-transferasa de carnitina 1A, en adultos que estuvieron expuestos durante su gestación a la hambruna que se le impuso a la población holandesa durante la Segunda Guerra Mundial (Hunger Winter)61,62. Los resultados evidenciaron que aquellos individuos cuyas madres fueron expuestas a la hambruna durante la etapa peri-concepcional presentaron cambios significativos en la metilación de estos genes respecto de sujetos controles. Este efecto sobre la metilación no estuvo presente en individuos a los cuales la hambruna afectó exclusivamente al término de su gestación. En conjunto estos antecedentes demuestran el carácter estímulo y tiempo-específico de las modificaciones epigenéticas, así como su proyección a largo plazo.
Adicionalmente, un creciente número de estudios ha revelado la presencia de marcadores epigenéticos, principalmente relacionados con la susceptibilidad a desarrollar diabetes tipo 2 en modelos animales de RCIU a través de la programación de la función hepática63,64 y pancreática65, así como el metabolismo en el musculoesquelético66. Así mismo, existe contundente evidencia de la función clave que tienen los mecanismos epigenéticos en el desarrollo de cáncer. En cambio, respecto al sistema vascular e inmune, cuyos desarrollos se encuentran altamente influenciados por mecanismos epigenéticos67–69, existe una evidencia más limitada.
Este artículo es la introducción a una serie de 5 revisiones en torno a la participación de los mecanismos epigenéticos en el desarrollo de enfermedades crónicas (i.e. cardiovasculares, metabólicas, asma/alergias y cáncer) y su relación con el origen de dichas enfermedades en etapas tempranas del desarrollo. El objetivo de esta serie es mostrar el estado actual de esta área de la investigación, y presentar los desafíos e interrogantes futuros en los cuales la pediatría tiene un papel preponderante, desarrollando estrategias para la prevención, detección precoz y seguimiento.
FinanciaciónFinanciado por Fondecyt Regular 1120928 (PC), 1130277 (RU), 1130801 (BJK) y 1141195 (JAC).
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
Esta actualidad forma parte de un ciclo de 5 actualidades consecutivas sobre el tema de Epigenética, a ser publicados en los números de 1 a 5 Vol. 87 de Revista Chilena de Pediatría 2016.
