




El conocimiento sobre los mecanismos que subyacen el desarrollo de la osteoporosis en el anciano ha avanzado de manera llamativa en los últimos años. Tras una brusca pérdida inicial de masa mineral ósea en el periodo perimenopáusico sigue una pérdida más progresiva y gradual que también ha sido vista en hombres. Esta pérdida inicial de masa ósea es debida a un aumento significativo de la resorción ósea. Con el envejecimiento existe además una reducción importante en la formación ósea que se debe, fundamentalmente, a que en la médula la osteoblastogénesis pasa a un segundo plano cediendo a la adipogénesis su papel principal. En este trabajo se revisa la última evidencia en la fisiopatología de la osteoporosis senil resaltando los mecanismos de acción de los tratamientos disponibles. Se han considerado también futuros potenciales tratamientos que incluyen nuevos abordajes terapéuticos basados en la fisiopatología de la osteoporosis en el anciano, y más precisamente, en la potencial reversibilidad de la adipogénesis.
Knowledge of the mechanisms underlying the development of osteoporosis in the elderly has advanced greatly in the past few years. After an initial sudden loss of bone mineral mass in the peri-menopausal period there follows a more progressive and gradual loss that has also been seen in men. This initial drop in bone mass is due to a significant increase in bone resorption. There is also a significant reduction in bone formation with age that is mainly due to osteoblastogenesis in the bone marrow passing to a second plane, transferring its main role to adipogenesis. In this article, the latest evidence on the pathophysiology of senile osteoporosis is reviewed, highlighting the mechanisms of action of available treatments. Potential future treatments are also considered, which include new therapeutic approaches based on the pathophysiology of osteoporosis in the elderly, mainly on the potential reversibility of the adipogenesis.
La osteoporosis se define como una enfermedad caracterizada por la pérdida de masa ósea junto con un deterioro de la microarquitectura que incrementa la fragilidad del hueso y le predispone a sufrir fracturas1. El incremento en la incidencia de fracturas osteoporóticas en mayores de 65 años se acompaña de un impacto catastrófico en la discapacidad y en la mortalidad2. Por ello, la osteoporosis es un importante problema de salud pública que afecta fundamentalmente a los ancianos.
La combinación de factores genéticos y nutricionales, así como la actividad física y el recambio óseo, determinan la cantidad de masa ósea y por lo tanto la resistencia del hueso3. Después de alcanzar el pico de la masa ósea durante la tercera década de la vida se produce un declinar progresivo de aproximadamente un 0,5% por año lo que es considerado como un cambio «fisiológico» relacionado con la edad4. Esta progresiva pérdida de masa ósea por sí sola no predispone necesariamente a sufrir fracturas. Sin embargo, múltiples factores, incluyendo los mencionados previamente, en combinación con esta pérdida de masa ósea incrementan el riesgo de fracturas en muchos individuos5.
El esqueleto está organizado en 2 compartimentos: periférico y axial. El esqueleto periférico, o cortical, constituye el 80% de la masa esquelética y está compuesto fundamentalmente por placas compactas (laminares) organizadas alrededor de canales centrales de los que se nutren. Los huesos largos están constituidos fundamentalmente por hueso cortical el cual encierra en su parte central la médula ósea6. El esqueleto axial, o central, está compuesto de hueso trabecular (esponjoso) en un 70% de su volumen y en un 35% de su peso aproximadamente. El hueso trabecular es un panal formado por líneas verticales y horizontales (trabéculas) entre las cuales se encuentra la médula ósea. La metáfisis de los huesos largos también contiene hueso trabecular, pero en el adulto carece de médula roja. Debido a que el hueso trabecular está en contacto directo con la médula ósea, la proximidad a estos componentes celulares determinará una respuesta más rápida y más intensa del hueso trabecular en la tasa de remodelación ósea7.
Existen 2 procesos fisiopatológicos que pueden producir una importante pérdida de masa ósea. El primero, conocido como osteoporosis posmenopáusica, es el resultado de la deprivación estrogénica y afecta primordialmente al hueso trabecular. Este tipo de osteoporosis afecta mayoritariamente a mujeres y se asocia frecuentemente a fracturas vertebrales y de radio8. Por el contrario, un segundo tipo de osteoporosis, conocida como osteoporosis senil, afecta fundamentalmente al hueso cortical predisponiendo tanto a mujeres como a hombres ancianos a sufrir fracturas de cadera9. Aunque el modelo unitario para la osteoporosis mantiene que la deficiencia estrogénica tiene un papel estelar en la fisiopatología de ambos tipos de osteoporosis, nuestra revisión dará mayor importancia a los mecanismos que subyacen la osteoporosis senil, que según nuestra hipótesis es el resultado de cambios producidos tanto en la celularidad del hueso como en las respuestas a factores hormonales y nutricionales. Destacaremos también los abordajes terapéuticos actuales y futuros en función de los cambios celulares asociados con la deprivación estrogénica y con el envejecimiento.
El proceso de remodelación del huesoEl recambio óseo es un ciclo continuo de destrucción y renovación de hueso como consecuencia de la acción conjunta de las células formadoras de hueso (osteoblastos) y de las células de resorción ósea (osteoclastos). Los objetivos de este proceso son corregir las microlesiones que sufre el hueso y adaptar la forma y la densidad del mismo en función de las fuerzas mecánicas y de los patrones de uso a los que es sometido. Ambos tipos de células están presentes en unas áreas denominadas unidades de remodelado óseo o unidades básicas multicelulares (UBM). El remodelado óseo comienza con la destrucción del hueso viejo por los osteoclastos seguido por el depósito de osteoide (hueso no mineralizado) por los osteoblastos10. Posteriormente, la matriz orgánica extracelular es mineralizada.
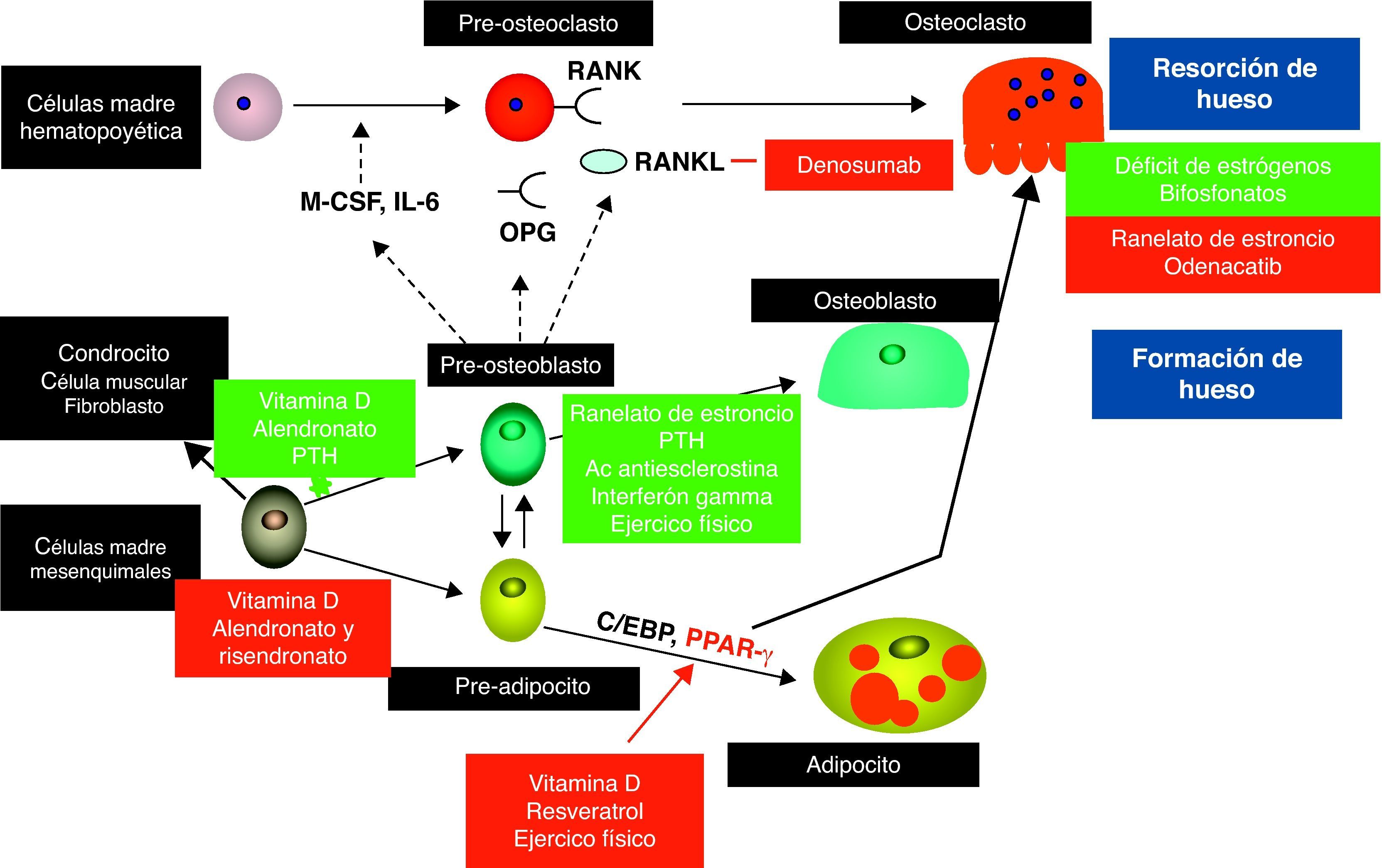
Los osteoclastos son células monocíticas hematopoyéticas que comparten un precursor común con los macrófagos11. Su membrana tiene una parte ondulada mediante la que se adhiere fuertemente al hueso encerrando una superficie ósea sobre la que tendrá lugar la resorción. La acidificación de este compartimento extracelular dará lugar a la desmineralización del hueso seguida por la activación de las cisteína proteasas, especialmente la catepsina K, que degradan la matriz orgánica12. Los osteoblastos, en cambio, son células fibroblásticas que se originan de precursores estromales en la médula ósea. Estas células tienen la capacidad de formar nuevo osteoide y estimular su mineralización13. El remodelado óseo está regulado por múltiples factores como hormonas (por ejemplo, estrógenos, hormona paratiroidea, vitamina D), interleucinas (por ejemplo, IL-1, IL-6, IL-11), citocinas (TNF-α) y factores de crecimiento (proteínas morfogenéticas del hueso)11. Todo este proceso requiere una estrecha comunicación entre el osteoblasto y el osteoclasto. Los osteoblastos responden a estímulos externos e internos tanto produciendo factor estimulante de colonias de macrófagos (M-CSF) como expresando en su membrana ligandos de receptor activados para el factor nuclear kB (RANKL), los cuales son factores indispensables para la osteoclastogénesis12; RANKL se une a su receptor (RANK) el cual se expresa en la membrana del osteoclasto y de sus precursores. La unión entre RANK y su ligando estimula la diferenciación y la activación del osteoclasto e impide su muerte celular14. Al mismo tiempo, este proceso es regulado por un receptor señuelo conocido como osteoprotegerina (OPG) el cual es producido por los osteoblastos e inhibe los efectos de la unión15. Muchos factores estimulan la expresión de RANKL como son la PTH, vitamina D, citocinas, interleucinas, prostaglandinas y tiazolidinedionas. Por contra, los estrógenos, el TGF-ß y la fuerza mecánica inhiben la expresión de RANKL. Recientemente, la señal producida por las efrinas ha sido implicada como parte importante en el acoplamiento osteoclasto-osteoblasto16. Esta comunicación celular es bidireccional e incluye al ligando transmembrana conocido como efrina B2, el cual es expresado por los osteoblastos. Esta señal limita la actividad del osteoclasto a la vez que estimula la diferenciación del osteoblasto16. Otros factores liberados por el osteoclasto desde la matriz ósea donde se produce la reabsorción modulan la formación osteoblástica y su actividad; éstos incluyen TGF-ß, proteínas morfogénicas del hueso, factor plaquetario derivado del hueso y lectina inhibidora de osteoclastos (OCIL).
Si el ciclo de la remodelación fuese completamente eficiente, nunca se perdería o ganaría hueso. Cada unidad de remodelación ósea renovaría completamente la parte de hueso que inicialmente reabsorbió. Sin embargo, la remodelación, como la mayoría de los procesos biológicos, no es completamente eficiente. De hecho, aunque este desequilibrio es minúsculo para una simple unidad de remodelación, produce una importante disminución en la masa total de aproximadamente 0,5% al año, dando lugar a una pérdida progresiva de masa ósea relacionada con la edad9.
Después de completar su función inicial, las células de la unidad de remodelación ósea toman diferentes destinos. Los osteoclastos mueren por apoptosis (o muerte celular programada) y son fagocitados in situ17. En cambio, los osteoblastos pueden tomar diferentes caminos; pueden convertirse en células de revestimiento (o células lineales) migrando a una nueva unidad ósea, pueden quedar atrapados en el osteoide convirtiéndose en osteocitos o, por último, morir por apoptosis9. La predominancia de cualquiera de estas opciones determinará la cantidad de osteoblastos disponibles en la unidad de metabolismo óseo y secundariamente la diferenciación y activación de los osteoclastos.
Pérdida de masa ósea relacionada con la edadLos cambios de la masa ósea con el envejecimiento son consecuencia de 2 procesos: la aposición del periostio que tiene lugar en la parte exterior del hueso y la resorción del endostio que tiene lugar en la parte interior del hueso. Curiosamente, tanto los hombres como las mujeres tienen un declinar similar en la resorción del endostio óseo, sin embargo, la aposición del periostio está menos afectada en los hombres18. Esto podría explicar por qué las fracturas vertebrales ocurren menos frecuentemente en hombres que en mujeres.
La pérdida de masa ósea relacionada con la edad es además consecuencia de cambios hormonales y celulares, tanto en cantidad como en función. Como se ha descrito previamente, la disminución de los niveles de hormonas sexuales en ambos sexos (aunque más significativamente en mujeres durante el periodo perimenopáusico) es seguida de un incremento en la formación y activación de los osteoclastos, debido tanto a un aumento de los valores de RANKL como a una disminución de la apoptosis de los mismos19. Una segunda hormona estrechamente ligada a los cambios relacionados con la edad en el hueso es la vitamina D. Existe una insuficiencia generalizada de vitamina D en el anciano, independiente de la latitud20. La reducción a la exposición solar y la disminución en la ingesta de comida rica en vitamina D contribuyen a esta hipovitaminosis20. Además, la capacidad de metabolizar vitamina D en la piel disminuye con el envejecimiento21. La hipovitaminosis D a menudo produce un hiperparatiroidismo secundario, el cual a su vez aumenta la resorción ósea por el osteoclasto22.
Además de los cambios hormonales, en el hueso se producen cambios celulares como alteraciones en la movilidad y diferenciación de las células madre mesenquimatosas (MSC)23. Esto da lugar a un aumento en el número de adipocitos con una disminución de los osteoblastos9. Tanto los osteoblastos como los adipocitos comparten los mismos precursores en la médula ósea, por lo tanto la adipogénesis se incrementa a expensas de la osteoblastogénesis24. Además, un aumento en la apoptosis reduce la vida media de los osteoblastos9. En resumen, los cambios celulares durante el envejecimiento óseo reducen el número de osteoblastos disponibles para la remodelación y formación ósea.
Aunque el proceso de pérdida de masa ósea relacionada con la edad es la consecuencia de cambios en la estimulación hormonal y en la celularidad ósea, una proporción de sujetos sólo la perderá de una manera «fisiológica», mientras que otro porcentaje sufrirá una «pérdida patológica de masa ósea» dando lugar a la osteoporosis. Aunque la dieta, la actividad física y la genética tienen un papel importante en la aceleración de esta pérdida de masa ósea, probablemente haya otros factores hormonales y moleculares que todavía no se hayan descubierto.
Envejecimiento y huesoAunque se sabe mucho sobre los potenciales mecanismos de pérdida de masa ósea relacionada con la edad, el enlace entre el proceso de envejecimiento normal y la osteoporosis senil permanece incierto. En un modelo de ratón con deficiencia en la reparación y transcripción de ADN, de Boer et al25 demostraron que el envejecimiento acelerado se acompañaba de cifosis y osteoporosis. Ratones sometidos a estrés oxidativo presentaron un fenotipo óseo similar26. Además, la preservación de la actividad de la telomerasa, que habitualmente se reduce con el envejecimiento y en las células senescentes, aumenta la supervivencia del osteoblasto y la generación de hueso lamelar27.
Probablemente la más interesante evidencia de un enlace entre envejecimiento y osteoporosis senil es la reciente descripción de mutaciones de la lamina A/C en el síndrome de progeria Hutchinson-Gilford28. Los pacientes con este síndrome tienen osteoporosis severa y cambios compatibles con envejecimiento óseo29, los cuales son similares a los encontrados en modelos de ratón sin lamina A/C que también tenían características de progeria30. Los osteoblastos envejecidos tienen disminuida la expresión de lamina A/C31 y alteraciones en la expresión de lamina A/C in vivo inducen una disminución en la actividad osteoblástica y osteocítica30 y alteraciones en la adipogénesis compatibles con lipodistrofia y redistribución grasa32. Aunque se requiere mayor investigación, la regulación de la expresión de la lamina A/C podría convertirse en un importante campo de investigación para la explicación de algunos de los mecanismos moleculares que subyacen en la osteoporosis senil.
Grasa y hueso: una extraña parejaEl hecho predominante en la osteoporosis senil es la acumulación de grasa en la médula ósea a expensas de la osteoblastogénesis33. Esta acumulación parece ser independiente de los estrógenos, ya que la grasa de la médula ósea aparece incluso cuando los niveles de estrógenos están todavía normales durante la tercera y cuarta décadas de la vida34. Además, ratones sin receptores estrogénicos no presentaron mayor adiposidad en su médula ósea que los ratones salvajes35. Por lo tanto, el envejecimiento per se, independientemente de los cambios hormonales, parece contribuir de manera significativa a la adipogénesis de la médula ósea, planteando la posibilidad de que la osteoporosis senil sea un tipo de enfermedad lipotóxica36. De hecho, se ha sugerido que la pérdida de masa ósea relacionada con la edad representa la «obesidad del hueso»33.
El papel de grasa de la médula ósea todavía permanece incierto. Aunque pudiera simplemente ocupar espacio que queda vacío tras la disminución de la hematopoyesis y la reducción de la masa trabecular, podría también desempeñar un importante papel patológico. Los adipocitos de la médula ósea ejercen un efecto tóxico en los osteoblastos37. Cocultivos de adipocitos y osteoblastos muestran que los adipocitos inhiben la actividad y la supervivencia de los osteoblastos, posiblemente debido a la liberación de adipocinas y ácidos grasos por el creciente número de adipocitos en la médula38. Datos recientes obtenidos en humanos tratados con tiazolidinedionas (rosiglitazona) apoyan el papel de la grasa de la médula ósea en la osteoporosis senil. La rosiglitazona induce la expresion de PPARγ2, un importante factor de transcripción que incrementa la adipogénesis medular asociada a la edad39. Estudios epidemiológicos revelan que los sujetos tratados con rosiglitazona no sólo sufren una importante pérdida de masa ósea40, sino que también tienen un mayor riesgo de fractura41. Sin embargo, la inhibición de la actividad de PPARγ, aunque reduce la acumulación de grasa en la médula del ratón diabético, no previene la pérdida de masa ósea42. En resumen, la grasa de la médula ósea induce cambios en la celularidad, especialmente en la línea osteoblástica que podrían explicar algunos de los cambios observados en la osteoporosis senil.
Mecanismos celulares del tratamiento de la osteoporosis: presente y futuroBasándose en los cambios celulares descritos en la osteoporosis senil (fig. 1), los objetivos terapéuticos deberían incluir la regulación de la actividad osteoclástica y por tanto del recambio óseo, el aumento en el número de osteoblastos disponibles para construir hueso nuevo y, finalmente, la regularización de la adipogénesis en la médula ósea43. Aunque muchos de los tratamientos actualmente disponibles son antirresortivos, que suprimen la actividad osteoclástica, hay evidencia reciente de que algunos de ellos pueden ejercer un efecto anabólico mediante la estimulación de la actividad osteoblástica y, en algunos casos, induciendo la diferenciación de adipocitos a osteoblastos. Además, algunos abordajes no farmacológicos también han demostrado alterar la celularidad del hueso.
")
Interacciones entre las diferentes poblaciones celulares en el hueso. Los osteoblastos regulan la diferenciación y actividad de los osteoclastos a través del sistema RANK/RANKL/OPG. Las células madre mesenquimatosas son los precursores de los osteoblastos. Con el envejecimiento, más células mesenquimatosas se diferencian en adipocitos los que a su vez estimulan los osteoclastos en un mecanismo que permanece poco claro.
La figura muestra los mecanismos de acción de los tratamientos para la osteoporosis desde el punto de vista celular. Los cuadros verdes indican estimulación mientras que los cuadros rojos significan inhibición. (Adaptada de Rosen et al. Nat Med. 2010.)
Los osteocitos son las principales células mecanosensoras del hueso, sin embargo, los mecanismos por los que responden a la actividad física o a la carga intermitente no están del todo esclarecidos. Entre las posibles vías de actuación, destaca el papel de la esclerostina, una proteína que inhibe la proliferación y la diferenciación de las células osteoblásticas y disminuye su esperanza de vida estimulando la apoptosis. La inhibición de la esclerostina producida por el ejercicio físico favorece, por lo tanto, la formación ósea44. Es decir, los osteocitos y los osteoblastos detectan la tensión mecánica estimulando su actividad y supervivencia. A su vez, debido a que la actividad de los osteoblastos y los osteoclastos está fuertemente acoplada, probablemente el efecto del estrés tenga un impacto en la actividad del osteoclasto y en la resorción ósea45. Así, el ejercicio, además de producir una mayor densidad de hueso, reduce la tasa de pérdida de masa ósea relacionada con la edad46.
El ejercicio físico ha demostrado beneficio tanto para prevenir el desarrollo de osteoporosis como para prevenir y tratar sus complicaciones47. Este efecto se debe, no sólo a la acción directa sobre el hueso, sino también a los cambios que se producen en el músculo: aumento de fuerza, mejora de equilibrio y disminución del número de caídas, principal factor de riesgo para las fracturas47. Está bien establecido que en los pacientes ancianos los ejercicios de carga intermitente mejoran la densidad mineral ósea48, sin embargo los efectos sobre la fuerza del hueso en el anciano no han sido bien evaluados hasta el momento49. Por todo esto, los programas de ejercicio para pacientes con osteoporosis deberían incluir ejercicios de soporte de peso, de equilibrio y de fortalecimiento muscular47.
Es de destacar que las vibraciones de cuerpo completo de baja frecuencia han demostrado mejorar la densidad mineral ósea y el equilibrio en mujeres posmenopáusicas50. Sin embargo, todavía no se han realizado estudios en mayores de 80 años.
Calcio y vitamina DEn sujetos ancianos la disminución en la absorción de calcio y los niveles reducidos de vitamina D dan como resultado valores elevados de PTH, manteniendo las concentraciones de calcio en plasma a expensas del hueso22. Por esto, clásicamente la sustitución con calcio y vitamina D constituye el primer eslabón en el tratamiento farmacológico de la osteoporosis. En un reciente metaanálisis se demostró que los suplementos de calcio aislados (sin vitamina D asociada) aumentan en un 30% el riesgo de infarto agudo de miocardio51. Estos hallazgos, junto con el escaso beneficio observado tras el tratamiento con calcio con o sin vitamina D en la prevención de fracturas52, replantean la indicación de este tratamiento, siendo aconsejable, hasta que aparezcan nuevas evidencias, no suplementar con calcio a menos que se acompañe de otros tratamientos para la osteoporosis53.
La vitamina D tiene un efecto dual en la prevención de fracturas en el anciano aumentando la densidad mineral ósea y la fuerza muscular. La dosis pautada y los niveles de 25(OH)D alcanzados en sangre se relacionan directamente con la reducción obtenida tanto en el número de fracturas como de caídas54,55, siendo recomendable pautar siempre altas dosis de vitamina D, al menos 800 UI al día, para alcanzar niveles mínimos de 30 ng/ml. Tanto la prevención de caídas como de fracturas sigue aumentando hasta niveles de 44 ng/ml. Clásicamente se entendía que el mecanismo de acción de la vitamina D en el hueso era el efecto indirecto en la absorción intestinal de calcio con la consecuente normalización de la PTH56. Sin embargo, trabajos recientes han demostrado que la vitamina D tiene efectos más sofisticados en el hueso envejecido. En primero lugar, la vitamina D aumenta la formación de hueso en animales con envejecimiento acelerado induciendo la osteogénesis y la actividad osteoblástica57. En segundo lugar, la vitamina D previene la apoptosis del osteoblasto in vitro58. Y por último, inhibe la adipogénesis en la médula ósea y la expresión de PPARg2 en ratones viejos mediante la estimulación de genes osteogénicos59. Estos trabajos, junto con la evidencia clínica, apoyan la importancia de la vitamina D para mantener la masa ósea y la calidad del hueso en el tratamiento de la osteoporosis senil tanto por sí misma como complemento a los tratamientos antirresortivos.
Hormonas esteroideasEs bien conocido que en mujeres la pérdida de estrógenos tiene múltiples efectos. Primero, hay una disminución en la eficiencia de la absorción intestinal del calcio60. Segundo, la deficiencia de estrógenos permite directamente la reclusión de un mayor número de osteoclastos lo que por sí mismo también parece reabsorber el hueso con mayor eficiencia19. Además, la deficiencia estrogénica produce un aumento de la secreción y producción de citocinas que estimulan la osteoclastogénesis, la activación del osteoclasto y la resorción ósea19. La consecuente destrucción de trabéculas disminuye la superficie ósea disponible para nueva formación ósea por lo que elementos trabeculares enteros pueden ser totalmente destruidos. Tercero, debido a que los estrógenos estimulan la apoptosis del osteocito, la deficiencia estrogénica tiene como resultado una disminución de la células que típicamente responden a las fuerzas mecánicas para mantener el esqueleto61. Cuarto, la disminución en el nivel de estrógenos favorece la diferenciación de la célula madre a adipocitos a costa de la osteoblastogénesis62. Por todo ello, la sustitución estrogénica en la perimenopausia y en edades posteriores protege la masa ósea y protege significativamente frente a las fracturas, sin embargo, inquietudes por las consecuencias perjudiciales sobre la salud han reducido considerablemente su uso63.
El papel de los andrógenos sobre el hueso esponjoso está mediado principalmente por la aromatización local a estrógenos64. La deficiencia androgénica en los hombres induce pérdida de hueso esponjoso similar a la que produce la deficiencia estrogénica en mujeres posmenopáusicas64, contribuyendo al desarrollo de osteoporosis. El tratamiento de la deficiencia franca de andrógenos con terapia sustitutiva es beneficioso para el hueso65,66 y para el músculo67, de hecho, la sustitución hormonal en pacientes con hipogonadismo se plantea como uno de los posibles tratamientos de la fragilidad68. Sin embargo, los efectos de la sustitución androgénica en la incidencia de fracturas y caídas, así como los efectos sobre otros tejidos, la calidad de vida y la supervivencia en el anciano se desconocen.
BifosfonatosLos bifosfonatos como el alendronato, el risendronato, el ibandronato y el ácido zoledrónico, regulan la reabsorción del hueso reduciendo el número de osteoclastos en la UBM e induciendo su apoptosis69. El mecanismo de acción de los bifosfonatos incluye la supresión de la capacidad de los osteoclastos de reabsorber el hueso debido a la modificación de su forma, la pérdida de su capacidad enzimática y la disminución de su supervivencia70. El resultado es una reducción importante en el recambio óseo que, secundariamente, aumenta la mineralización de las osteonas preformadas70. Es interesante que, a pesar de una reducción en el recambio óseo, la masa ósea se incrementa durante el tratamiento con bifosfonatos. Esto podría explicarse por un efecto anabólico o un efecto de los bisfosfonatos en la mineralización secundaria como muestran publicaciones recientes en las que el alendronato estimula la diferenciación osteoblástica a la vez que inhibe la adipogénesis in vitro71,72 y otra, del mismo grupo, en la que el risendronato disminuye la grasa de la médula ósea in vivo73. Aunque este aumento de la masa ósea está asociado, en mujeres posmenopáusicas, a una reducción en la tasa de fracturas tanto vertebrales como no vertebrales70, la evidencia sobre el efecto de los bifosfonatos en la prevención de fracturas en la población anciana es relativamente limitada74. Sin embargo, estudios recientes han mostrado que el ácido zoledrónico anual intravenoso reduce significativamente las fracturas vertebrales y no vertebrales (incluida fractura de cadera) en mujeres ancianas (mayores de 75 años)75 y reduce significativamente las fracturas vertebrales y no vertebrales (pero no necesariamente fractura de cadera) así como la mortalidad (∼ 28%) en pacientes ancianos que ya han sufrido fractura de cadera76.
Recientemente, el tratamiento a largo plazo con bifosfonatos (fundamentalmente alendronato) se ha asociado a fracturas atípicas de fémur. Sin embargo, el beneficio en la prevención de fracturas supera con creces el riesgo de estas fracturas atípicas, por lo que la abstención terapéutica por esta causa no está justificada77.
Ranelato de estroncioEl ranelato de estroncio es una molécula con un efecto dual: inhibe la resorción y estimula la formación ósea por un efecto directo sobre el osteoclasto y el osteoblasto respectivamente, lo que se manifiesta con cambios en los marcadores óseos78. Los 2 estudios principales, el Spinal Ospeoporosis Therapeutic Intervention (SOTI79) y el TReatment Of Peripheral Osteoporosis (TROPOS80), han demostrado una reducción en la incidencia de fracturas. Un análisis a partir de datos de estos 2 trabajos muestra, en ancianas mayores de 80 años, una reducción significativa del riesgo de fractura vertebral, no vertebral y clínica al año y a los 3 años del tratamiento81. El fármaco fue bien tolerado y con un perfil de seguridad equiparable al de las pacientes más jóvenes.
Tratamiento anabólicoActualmente, la hormona paratiroidea y su análogo la teriparatida (PTH [1-34] recombinante) son los únicos tratamientos anabólicos aprobados para la osteoporosis82. Dosis intermitentes diarias de PTH inducen la diferenciación osteoblástica, inhiben la adipogénesis y suprimen la apoptosis del osteoblasto82. Esto se contrapone con la resorción ósea que está causada por una persistente elevación de la hormona paratiroidea en determinadas enfermedades como el hiperparatiroidismo primario y algunos tumores. La teriparatida está indicada en mujeres posmenopáusicas y en hombres con osteoporosis severa (T-score ≤ –3,5 o T-score < –2,5 asociado a fracturas recurrentes) o en pacientes con osteoporosis establecida inducida por glucocorticoides que requieren tratamiento esteroideo a largo plazo83,84. La teriparatida en mujeres mayores de 75 años que viven en la comunidad aumenta la densidad mineral ósea, reduce las fracturas vertebrales y no vertebrales y es tan segura como en mujeres jóvenes84. Las limitaciones para la administración de PTH en ancianos incluyen el coste, el modo de administración (subcutáneo) y la necesidad de un paciente motivado y cognitivamente intacto o de un cuidador.
DenosumabRecientemente se ha aprobado el denosumab, un anticuerpo monoclonal que se une al ligando de receptor activado para el factor nuclear κB (RANKL) impidiendo su unión al receptor RANK85. De esta forma inhibe el desarrollo y la actividad de los osteoclastos, disminuyendo la resorción del hueso e incrementando la densidad ósea. En definitiva, el denosumab simula la acción de la osteoproteregina85. El denosumab tiene una vida media larga (1-1,5 meses) y administrado en dosis única subcutánea induce una rápida (12h) y prolongada (más de 6 meses) inhibición de la resorción ósea en mujeres posmenopáusicas86. El estudio FREEDOM, que evalúa el efecto del denosumab en el riesgo de fractura en mujeres posmenopáusicas con osteoporosis, demuestra una disminución significativa de fracturas vertebrales, no vertebrales y de cadera85.
Perspectivas futuras para el tratamiento de la osteoporosis en el ancianoDurante los últimos años han surgido nuevas opciones terapéuticas para el tratamiento de la osteoporosis en el anciano, algunas de ellas muy recientes. Pero todavía hay nuevas moléculas en fase de desarrollo, entre las que cabe destacar:
- •
La cathepsina K es una proteasa lisosomal que es expresada por los osteoclastos y se encarga de degradar el colágeno. El inhibidor de la cathepsina K (odenacatib) ha demostrado disminuir la resorción ósea sin disminuir sustancialmente la formación de hueso, lo que conlleva a un aumento de la densidad mineral ósea87. La eficacia antifractura del odenacatib está siendo probada actualmente en un estudio fase III.
- •
La esclerostina es una proteína producida por los osteocitos que inhibe la proliferación y la diferenciación de las células osteoblásticas y disminuye su esperanza de vida estimulando la apoptosis. Los anticuerpos antiesclerostina han demostrado en estudios preclínicos un marcado efecto anabólico88.
- •
El interferón gamma, una molécula producida por las células immunitarias y MSC ha sido recientemente descrito como un posible tratamiento anabólico de la osteoporosis. El efecto de bajas dosis de interferón gamma en el hueso es muy similar al de la PTH ya que a baja dosis aumenta la osteoblastogénesis e inhibe la adipogénesis mientras que a altas dosis aumenta la osteoclastogénesis89.
- •
PPARγ favorece la diferenciación a adipocito de la célula mesenquimatosa a costa de la osteoblastogénesis, por lo que diferentes factores inhibidores de este factor se han convertido en diana como posibles opciones terapéuticas de la osteoporosis en el anciano39. Entre otros futuros tratamientos cabe destacar el resveratrol, que deriva de la piel de la uva y subyace la paradoja del «vino tinto»90 y el ejercicio físico91.
El hueso es un tejido activo que no sólo actúa como soporte corporal, sino que también es un órgano metabólico con múltiples interacciones celulares y hormonales. La regulación de la diferenciación, la activación y al acoplamiento celular en el envejecimiento óseo probablemente repercuta en el riesgo de fractura de los pacientes ancianos. La elección del tratamiento óptimo debería basarse en las características particulares de cada paciente y en un completo entendimiento de los mecanismos celulares implicados con el fin de proporcionar el tratamiento más efectivo. Aunque la mayoría de los tratamientos disponibles hoy en día actúan directamente en la regulación de la resorción del hueso, futuros trabajos deberían centrarse en el desarrollo de tratamientos anabólicos con el objetivo de preservar la cantidad y la calidad adecuada de masa ósea en el anciano.
FinanciaciónEl profesor Duque es consultor y ha recibido subvenciones de investigacion de las siguientes compañías: Novartis, Merck, Procter & Gamble, Servier.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Todos los trabajos de investigación por parte de los autores que han sido mencionados en este artículo han sido financiados con subvenciones del Australian National Health and Medical Research Council (NHMRC), el Canadian Institutes of Health Research (CIHR) y la Nepean Medical Research Foundation. La Dra. Alonso-Bouzón escribió este artículo como parte de su Visiting Scholarship en la Universidad de Sydney patrocinada por la Fundación del Hospital Getafe (Madrid, España).
