


Most of pathogenesis related (PR) proteins possess complicated structures; hence their active recombinant forms are usually produced in eukaryotic systems. In this study, we employed an insect cell line to express a recombinant form of a previously identified grape PR3 allergen categorised as class IV chitinase.
MethodsGrape chitinase cDNA was amplified by RT-PCR and inserted into pFastBacHTA using restriction enzymes. The recombinant pFastBacHTA was applied for the transformation of Escherichia coli DH10Bac cells. The purified recombinant bacmid was used for transfection of Sf9 cells. Finally, the IgE-immunoreactivity of purified recombinant protein was evaluated using grape allergic patient's sera. Moreover, polyclonal anti-6His-tag and monoclonal anti-chitinase antibodies were used for further assessment of recombinant protein.
ResultsSDS–PAGE analysis of the transfected Sf9 cells showed expression of a monomeric 25kDa and a dimeric 50kDa recombinant protein. Western blotting revealed considerable IgE reactivity of the recombinant protein with grape allergic patients’ sera. Furthermore, confirmatory assays showed specific reactivity of the recombinant protein with anti-His tag and anti-chitinase antibodies.
ConclusionThis study showed that, in contrast to E. coli, insect cells are suitable hosts for the production of a soluble and IgE-reactive recombinant form of grape class IV chitinase. This recombinant allergen could be used for component resolved diagnosis of grape allergy or other immunodiagnostic purposes.
Since two decades ago, component resolved diagnosis (CRD) has become a popular technique in allergy studies.1,2 In several studies, natural or recombinant forms of food/fruit allergens have been applied for CRD.3 However, since purification of natural allergens could be a costly procedure and sometimes may result in immunological inactivation of the allergens, recombinant technology has become the first method of choice for preparation of sufficient amounts of high quality homogenous proteins for CRD purposes. These recombinant proteins could be used for skin prick tests (SPT),2,4 development of allergen specific IgE immunoassays, and even allergy immunotherapy.5 However, the structural similarity of recombinant allergens and their natural counterparts is the basic requisite to ensure their equivalent immunological properties.6 Among many available expression systems, Escherichia coli (E. coli) strains are still a popular choice because of their low cost and high level expression properties.7,8 However, in the case of complex proteins/allergens which need different post translational modifications (PTM), they are not assumed as the best option. Usually incorrect disulphide bond formation, improper protein folding and lack of protein glycosylation are the main problems, which could be resolved by switching towards a eukaryotic expression system.7 Using methylotroph Pichia pastoris to obtain high level expression of recombinant protein is the second most popular approach; however, yeasts may also have limitations in the production of some complex proteins.9 Production of heterologous proteins in insect cells using baculovirus technology is another choice. This approach usually leads to expression of correctly folded and post-translationally modified soluble heterologous proteins.10–12
Despite the mentioned problems, to date most of the recombinant allergens have been produced in bacterial hosts. Refolding may partially improve the conformation of some misfolded recombinant proteins and retrieve their IgE reactivity.13 However, proper glycosylation may be necessary not only for suitable folding but also immunoreactivity of allergic epitopes, especially cross reactive carbohydrate determinants.14,15 Thus, choosing an appropriate vector and a suitable host is the prerequisite step for producing a functional recombinant allergen.16 Overall, such a goal could be better achieved by expression of allergens in eukaryotic systems.
The family of pathogenesis related (PR) proteins comprises approximately 25% of plant allergens, and they are also considered as the main allergens of fruits.17 PR proteins are highly expressed by plants in response to stress conditions such as infections and exposure to certain abiotics18. In several studies, they have been cloned and expressed as IgE reactive proteins in various hosts. Some studies show that they exhibit a better immunoreactivity when expressed in eukaryotic cells, as compared to bacterial hosts.19,20
Chitinases are glycosyl hydrolases that catalyse degradation of chitin, the second most abundant polymer in nature. They comprise one of the main groups of PR proteins. In some microorganisms these enzymes may have a nutritional profit. However, in plants they play a role in natural defence reactions against the invasion of pathogenic microorganisms.21 They also participate in mammalians’ innate immunity. An acidic chitinase is expressed in the gastrointestinal tract, as well as in lung and conjunctiva epithelial cells, while other types of chitinolytic enzymes may be found in phagocytic cells. Lately, an elevated level of acidic chitinase was demonstrated in lung and cornea epithelial cells and its association with TH2 inflammatory disease was confirmed. Moreover, it was shown that the chitinolytic activity of tears is a protective factor in the control of chitin-containing pathogens.22,23
Recently, molecular characterisation of grape (Vitis vinifera) allergens has revealed their identity as members of PR proteins.24–27 Class IV chitinase is one of the major allergens of this unique horticultural crop which is over-expressed during its ripening time.24 The current study was performed to produce this allergen in insect cells and E. coli expression systems and evaluate their immunoreactivity via grape allergic patients’ sera.
MethodsRNA extraction and cDNA synthesisThe ripe grape berries (V. vinifera cultivar sultana) were collected from Golmakan vineyard of the Iranian Ministry of Agriculture. Total RNA was isolated from grapes, using the efficient rapid method designed by Fort et al. with some minor modifications, especially in the DNA removal step.28 For the elimination of genomic DNA impurities, the extracted RNA was treated with DNase I (Fermentas, Lithuania) at 37°C for 30min, followed by a further 10min incubation at 70°C for enzyme inactivation. Reverse transcription was carried out by 2μg of grape total RNA using RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Lithuania) according to the manufacturer's instructions.
Cloning and expression of grape chitinase in baculovirus systemPrimer designCloning primers with specific overhangs were designed for the amplification of coding region of grape class IV chitinase, according to available gene bank sequences. A specific NcoI recognition site was considered to be inserted in the forward primer (5′-ATA CCA TGG CAC AGA ACT GTG GGT GTG CCT CAG-3′), and the reverse primer (5′-ATA CTC GAG TTA GCA AGT GAG GTT GTC ACC AGG TG-3′) contained a stop codon as well as a specific XhoI recognition site (enzyme recognition sites were underlined).
Amplification of the target geneThe target chitinase coding sequence was amplified by RT-PCR method. The PCR reaction was carried out in 20μl volumes containing 1μl of cDNA, 1× PCR reaction buffer, 1.5mM MgCl2, 10pM of each primer, 1mM dNTPs, and 2units of Ex-Prime Taq DNA polymerase (Genet Bio, Korea). A thermal cycler was programmed with an initial denaturation at 94°C for 5min followed by 35 cycles of 95°C for 45s, 57°C for 45s, 72°C for 1min; and a final elongation time of 10min at 72°C. The amplification was evaluated by agarose gel electrophoresis.
Construction and characterisation of recombinant donorThe RT-PCR product as well as donor pFastBacHTA plasmid (Invitrogen, CA, USA) were digested with NcoI and XhoI enzymes (Fermentas, Lithuania) (16h, 37°C) and following electrophoresis on 1% agarose gel, the final products were extracted by DNA purification kit (Bioneer, Korea). The purified double-digested products were ligated using T4 DNA ligase (16°C, overnight). The construct was transformed into competent E. coli DH5α cells (Invitrogen, CA, USA) using the Inoue procedure.26 The transformants were selected on LB agar plates supplemented with 100μg/ml Ampicillin (Amp). The chosen colonies were subcultured overnight in LB broth and the recombinant plasmid was purified using conventional miniprep alkaline lysis method and subjected to DNA sequencing using polyhedrin forward and SV40pA reverse universal primers.29,30
Construction and characterisation of recombinant bacmidCompetent E. coli DH10Bac (Invitrogen, CA, USA) cells, containing the bacmid genome and helper plasmid were transformed with 10ng of the purified recombinant donor plasmid using the Inoue procedure. Following the heat shock, 300μl of preheated SOC media was added and the microtube was incubated 4h on shaker at 37°C to allow for expression of the antibiotic resistance genes as well as transposition of the target gene. The microtube contents were diluted 1:10, 1:100, 1:1000 with SOC media and 50μl of each diluted sample was placed on fresh LB agar supplemented with 50μg/ml kanamycin, 7μg/ml gentamicin, 10μg/ml tetracycline, 100μg/ml X-gal and 40μg/ml IPTG as recommended by Luckow et al.12 Following 48h incubation in 37°C, the LacZα gene disrupted white colonies were selected and re-streaked as possible recombinants. Finally, the large white colonies were chosen for bacmid DNA isolation.
Extraction and analysis of recombinant bacmid DNAHigh molecular weight bacmid DNA was isolated by miniprep alkaline lysis method29,30 with some minor modifications. In details, selected white colonies were shaker-incubated (37°C, overnight) in LB broth media supplemented with 50μg/ml kanamycin, 7μg/ml gentamicin and 10μg/ml tetracycline. The cells were harvested by centrifugation at 8000×g for 1min. The pellet was thoroughly re-suspended in 200μl of cold re-suspension solution (25mM Tris–HCl pH 8.0, 50mM glucose, 10mM EDTA, 20μg/ml RNAse A), followed by 400μl of freshly prepared lysis solution (200mM NaOH, 1% SDS). The microtubes were slowly inverted three times and incubated at RT for 5min. Subsequently 300μl of 3M potassium acetate pH 5.5 was added and the microtubes were kept on ice for 10min. The E. coli genomic DNA and proteins were pelleted by centrifugation at 12,000×g for 10min at 4°C. The supernatant was added on 800μl of cold isopropanol and kept on ice for exactly 10min, to allow the precipitation of the bacmid DNA. The samples were centrifuged at 12,000×g for 15min at room temperature (RT) and the pellets were washed three times by 70% ethanol. The pellets were vacuum-dried and re-suspended in 20μl of TE buffer and checked by electrophoresis on 0.5% agarose gel. Moreover, the transposition of chitinase coding region to bacmid DNA was verified by PCR analysis using pUC/M13 universal primers. The amplification result was analysed on 1% agarose gel.
Insect cell transfection and baculovirus stock preparationSpodoptera frugiperda (Sf9) insect cells (National Cell Bank of Iran Code: C425; Pasture Institute, Tehran, Iran) were maintained as adherent monolayer cells in Grace's media supplemented with 10% FCS or alternatively 10% lactalbumin hydrolysate/yeast extract (Sigma–Aldrich, St. Louis, MO, USA) at 27°C in a humidified incubator until it reached 70–80% confluency. The extracted recombinant bacmid was transfected into grown Sf9 cells by lipofection. Briefly, 24-well tissue culture plates were seeded at a density of 60,000 cells per well with Sf9 cells in 2ml of Grace's complete media and incubated at 27°C overnight. The Baculoporter™ transfection reagent (Genlantis, San Diego, CA, USA) (1μl) and the purified recombinant bacmid DNA (4μg) were diluted separately in 125μl of serum and antibiotic free Grace's media; then mixed together, and incubated 20min in dark at RT to form lipid–bacmid complexes. The supernatant of the 24-well tissue culture plate was carefully aspirated and wells were washed twice with serum free media and substituted with 250μl of the lipid–bacmid mixture and incubated 4h at 27°C. Finally, the transfection mixture was replaced with 2ml of Grace's complete media and the plate was incubated at 27°C for 96h. At the late stage of infection, the cell culture supernatant was harvested as the passage 1 (P1) virus stock. To prepare the P2 virus stock, 100μl of P1 was used to infect 5×106 monolayer Sf9 cells. Following 72h incubation at 27°C the supernatant was collected and centrifuged at 2000×g for 5min and filtered through a 0.22μm membrane and stored at 4°C as recombinant baculovirus stock.
Expression of recombinant proteinThe protein expression was performed using P2 stock. Sf9 monolayer cells (5×106) were infected at a multiplicity of infection (MOI) of approximately 10 plaque-forming units per cell. The flasks were incubated at 27°C for 96h and finally the cells were pelleted by centrifugation at 2000×g for 5min. The harvested cells were washed twice with homogenisation buffer (10mM Tris–HCl pH 8, 25mM NaCl) and re-suspended in 100μl of the same buffer. The cells were lysed through three continuous freeze and thaw cycles and the lysate was clarified by centrifugation at 12,000×g for 5min and the supernatant was subjected to the nickel-chelating affinity chromatography and the soluble recombinant protein was extracted via native protein purification method.
Cloning and expression of grape chitinase in prokaryotic systemTo check if the bacterial systems could produce an IgE-reactive chitinase or not; the above-mentioned primers (with different overhangs) were used for cloning of grape class IV chitinase in bacterial system. A forward primer with NotI restriction site (5′-ATA GCG GCC GCA CAG AAC TGT GGG TGT GCC TCA G-3′) and a reverse primer with XhoI restriction site (5′-ATA CTC GAG GCA AGT GAG GTT GTC ACC AGG TG-3′) were exploited for directional cloning of RT-PCR product into pET21b+ cloning vector (Invitrogen, CA, USA). Briefly, after restriction enzyme digestion and ligation steps, the construct was transformed into competent E. coli TOP10 cells (Invitrogen, CA, USA). The recombinant plasmid was analysed by sequencing via universal T7 primers. The recombinant chitinase was expressed in E. coli BL21 Star (DE3) as well as E. coli Origami B cells, as previously described.31 The bacterial cells were lysed and the contents (supernatant and the pellet) were subjected to electrophoresis procedures to determine protein expression status. Moreover, the inclusion bodies were purified and solubilised in 6M guanidinium–HCl. Finally, the 6His-Tag bound proteins were separated by nickel-chelating affinity chromatography as recommended by manufacturer (Ni-NTA-agarose, Invitrogen, CA, USA). The oxidative refolding of the product was performed through step-by-step dialysing of the purified protein against 50mM Tris–HCl pH 8.0 containing decreasing amounts of DTT (20–0mM), to achieve appropriate three-dimensional structure of the protein.32,33
SDS–PAGE and Western blottingGrape proteins were fractionated and purified as previously described.24,34 Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) was carried out using 12.5% separating and 5% stacking gels. The protein bands from recombinant chitinase expressing Sf9 cell lysates, as well as grape natural chitinase and the recombinant forms produced in bacterial hosts were resolved and visualised by Coomassie blue staining or subjected to Western blotting as previously reported.24,34 For Western blotting, the resolved protein bands were transferred on polyvinylidene difluoride (PVDF) membrane. Afterwards, the protein free sites were blocked by 2% bovine serum albumin (4°C, overnight) and the membranes were alternatively incubated with grape allergic patients’ pooled sera, anti-6His-tag or monoclonal anti-class IV chitinase antibodies.
For evaluation of IgE-reactivity of the produced proteins, the membranes were incubated (4°C, overnight) with a 1:10 dilution of pooled sera from ten patients who suffered from mild to severe symptoms of grape allergy and demonstrated a positive skin prick test with grape crude extract as well as a positive Western blotting with natural grape class IV chitinase (patients demographic data, not shown). After a washing step, the membranes were incubated with biotinylated goat anti-human IgE (KPL, Gaithersburg, MD, USA) (1:2000, 2h, RT). Following another washing step, HRP conjugated streptavidin (BD Biosciences Pharmingen, USA) was used for recognition of anti-human IgE binding (1:30,000, 1h, RT).
For assessment of production of His-tag bound protein, the membranes were incubated with biotinylated monoclonal anti-6His-tag antibody (Roche, Germany) (1:2000, 2h, RT). After washing, the bound antibody was detected by 1:30,000 dilution of HRP conjugated streptavidin (1h, RT).
Detection of class IV chitinase was fulfilled by incubation of the membrane with a mixture of supernatants of anti-class IV chitinase monoclonal antibody producing hybridoma cells (1:100, 3h, RT).34 Binding of monoclonal antibodies was checked by biotinylated rabbit anti-mouse antibody (KPL, Gaithersburg, MD, USA) (1:3000, 2h, RT) followed by HRP conjugated streptavidin (1:30,000, 1h, RT). Finally, in all Western blotting assays, the Immunoreactivity signals were recorded by chemiluminescent method, as previously reported.34
ResultsMolecular cloning of chitinase coding cDNA in baculovirus systemThe A260/A280 and A260/A230 ratios of the purified RNA were 1.87 and 1.79, respectively; indicating successful purification of RNA from grape berries. Electrophoresis revealed considerable 18 S and 25 S bands with visible smear, indicating acceptable quality of RNA. RT-PCR amplification of grape cDNA resulted in a single band around 750bp which was directionally subcloned into the pFastBacHTA baculovirus transfer vector. DNA sequencing of the purified recombinant vector revealed that a 738bp fragment had been successfully cloned into the donor plasmid. This fragment showed 97% identity with a previously predicted sequence of grape (Accession No. XM_002273565.1) and 83% identity with the coding sequence of grape class IV chitinase (CHI4D, Accession No. XM_002275350.1) or endochitinase (VvChi4B, Accession No. U97522.1). Translation of the cloned sequence showed that the expressed protein also had approximately 90% similarity with the previously reported grape class IV chitinases. The recombinant donor plasmid was transformed into E. coli DH10Bac cells, and consequently the recombinant bacmid was selected by plating a serial dilution of the transformed cells on selection media. Transposition resulted in disruption of the LacZα gene and production of around ten white colonies which were double checked by re-striking on selective media. Serial dilution of the bacterial cells at the transposition step was very useful and after 48h incubation in 37°C, only plates with lower cell densities (1:100 and 1:1000 dilutions) contained distinct large white colonies. The other plates which had higher cell densities contained many small colonies, in which the white and blue colonies could not be distinguished from each other.
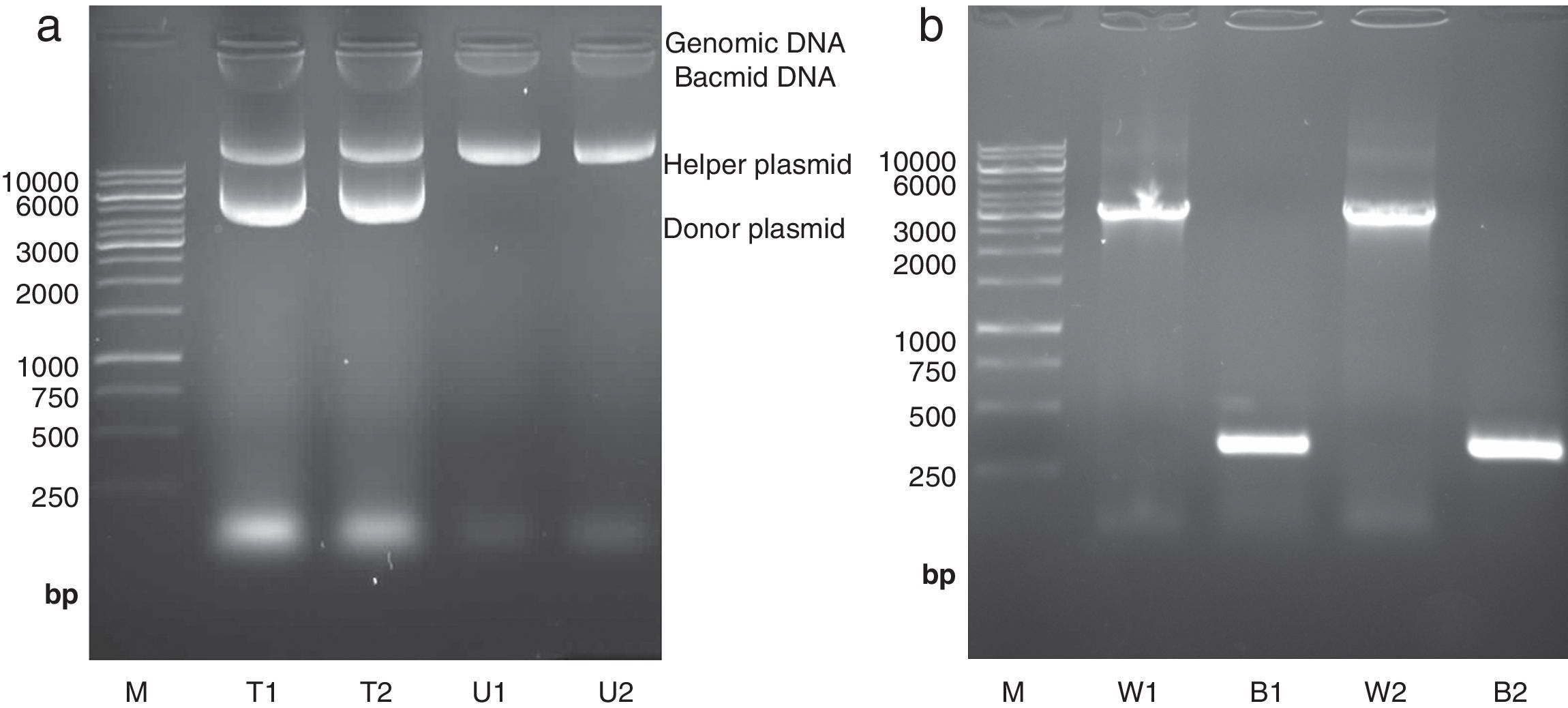
High molecular weight bacmid was purified from selected colonies – as well as blue colonies for control (Fig. 1a). The correct transposition of the recombinant bacmid was confirmed by conventional PCR, using pUC/M13 universal primers. As shown in Fig. 1b, when non-recombinant colonies were used as PCR template, a 300bp amplicon was observed; whereas, the recombinant bacmids resulted in amplification of an approximately 3200bp single band, consistent with the combined size of the mini-attTn7 (2430bp) and donor plasmid inserted fragment (738bp). The absence of the 300bp band in recombinant bacmids indicated that the recombinant bacmid was pure and free of contamination with wild type bacmid. No amplification was observed in a negative control microtube containing distilled water. Finally, one of the positive bacmids was selected for transfection of Sf9 cells.
 Isolation of high molecular weight bacmid by modified alkaline lysis method. The purified bacmids from two white colonies (i.e. transposition of target gene has occurred) (T1 and T2) as well as bacmids from untransformed DH10Bac colonies (U1 and U2) were run on 0.5% agarose in parallel to Fermentas 1kbp DNA marker (M). The purified bacmids also show some genomic DNA impurities. Moreover, the purified recombinant bacmids (T1 and T2) also contain an extra band which refers to the transformed donor plasmid (pFastBacHTA-Chitinase) with an approximately 6kbp size. (b) Analysis of recombinant bacmid DNA holding grape class IV chitinase by conventional PCR. Purified bacmids from two white (W1 and W2) or blue (B1 and B2) E. coli DH10Bac colonies used as template in PCR reaction with pUC/M13 primers and the amplicons were examined by agarose gel electrophoresis. The PCR reaction of bacmids from white colonies (supposed of cloning grape class IV chitinase) resulted in amplification of an approximately 3200bp band resembling appropriate transposition of the target gene. The PCR product from blue colonies showed amplification of a 300bp DNA, resembling a wild type AcMNPV bacmid. Fermentas 1kbp DNA ladder were used as marker (M) in both electrophoresis.")
Isolation and analysis of bacmid from E. coli DH10Bac cells. (a) Isolation of high molecular weight bacmid by modified alkaline lysis method. The purified bacmids from two white colonies (i.e. transposition of target gene has occurred) (T1 and T2) as well as bacmids from untransformed DH10Bac colonies (U1 and U2) were run on 0.5% agarose in parallel to Fermentas 1kbp DNA marker (M). The purified bacmids also show some genomic DNA impurities. Moreover, the purified recombinant bacmids (T1 and T2) also contain an extra band which refers to the transformed donor plasmid (pFastBacHTA-Chitinase) with an approximately 6kbp size. (b) Analysis of recombinant bacmid DNA holding grape class IV chitinase by conventional PCR. Purified bacmids from two white (W1 and W2) or blue (B1 and B2) E. coli DH10Bac colonies used as template in PCR reaction with pUC/M13 primers and the amplicons were examined by agarose gel electrophoresis. The PCR reaction of bacmids from white colonies (supposed of cloning grape class IV chitinase) resulted in amplification of an approximately 3200bp band resembling appropriate transposition of the target gene. The PCR product from blue colonies showed amplification of a 300bp DNA, resembling a wild type AcMNPV bacmid. Fermentas 1kbp DNA ladder were used as marker (M) in both electrophoresis.
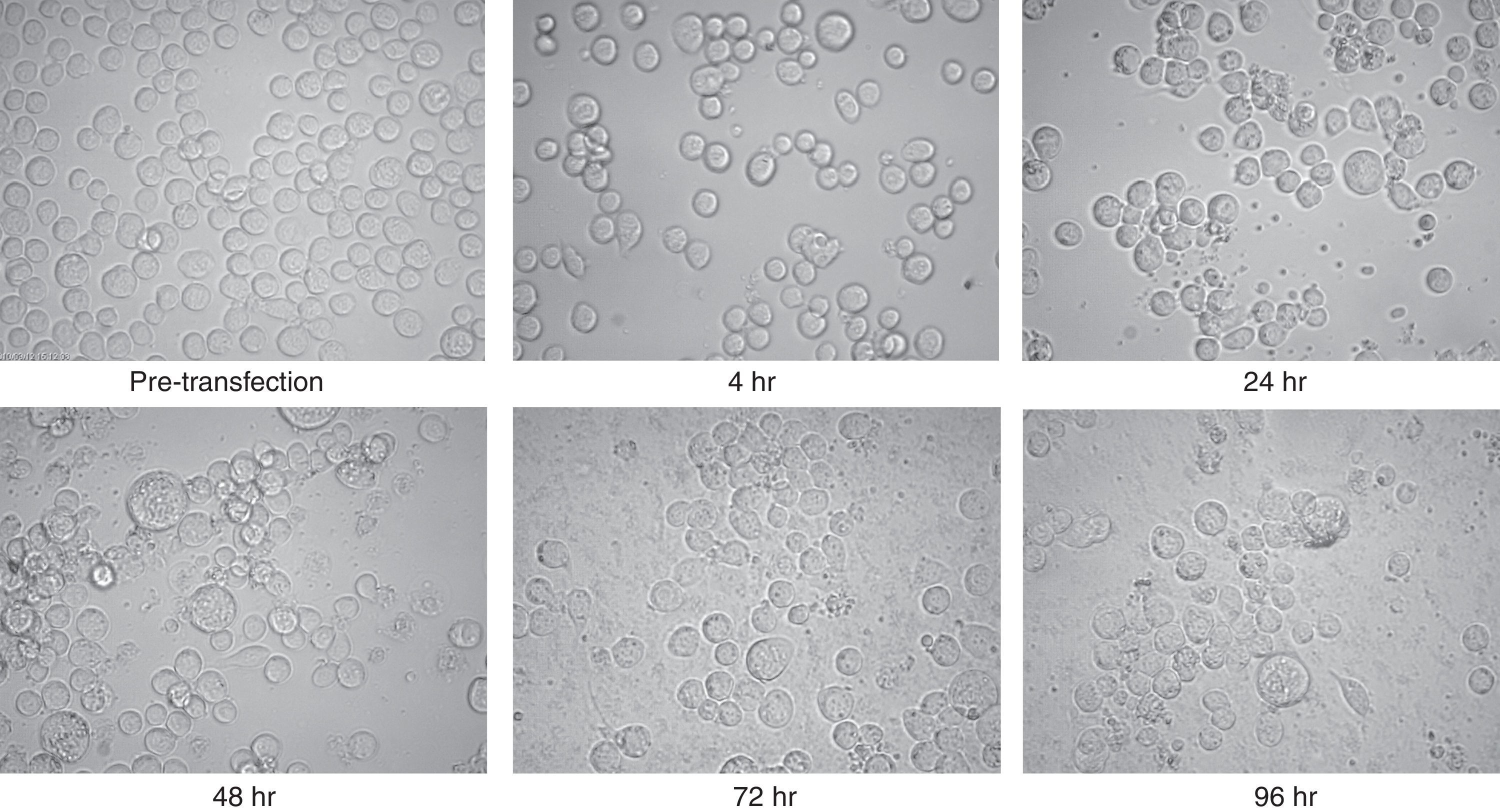
The purified recombinant bacmid was successfully used for transfection of Sf9 cell. The growth of Sf9 cells was diminished after transfection, and viral inclusion bodies as well as cell debris could be observed around 48h (Fig. 2). The baculovirus rich supernatant was applied for transfection and expression of recombinant protein of fresh Sf9 cells. Finally, the whole cell lysates were analysed by SDS–PAGE and Western blotting. In contrast to the high level expression in bacterial host, Coomassie blue staining of the SDS–PAGE gels showed two faint bands with an apparent molecular weight of 25 and 50kDa in cell lysates. These bands were absent in mock-infected cells.
 were captured. Note the increment of cell diameter and granular appearance of transfected cells and also their lysis at the final steps. The increased amount of cell debris and decreased cell count were obvious in late hours post transfection. There was no significant difference between the appearances of the cells which were infected with wild type or recombinant bacmid.")
Microscopic view of Sf9 cells after transfection with purified bacmid via lipofection. Cells from pre-transfection as well as several hours post-transfection (4, 24, 48, 72, and 96h) were captured. Note the increment of cell diameter and granular appearance of transfected cells and also their lysis at the final steps. The increased amount of cell debris and decreased cell count were obvious in late hours post transfection. There was no significant difference between the appearances of the cells which were infected with wild type or recombinant bacmid.
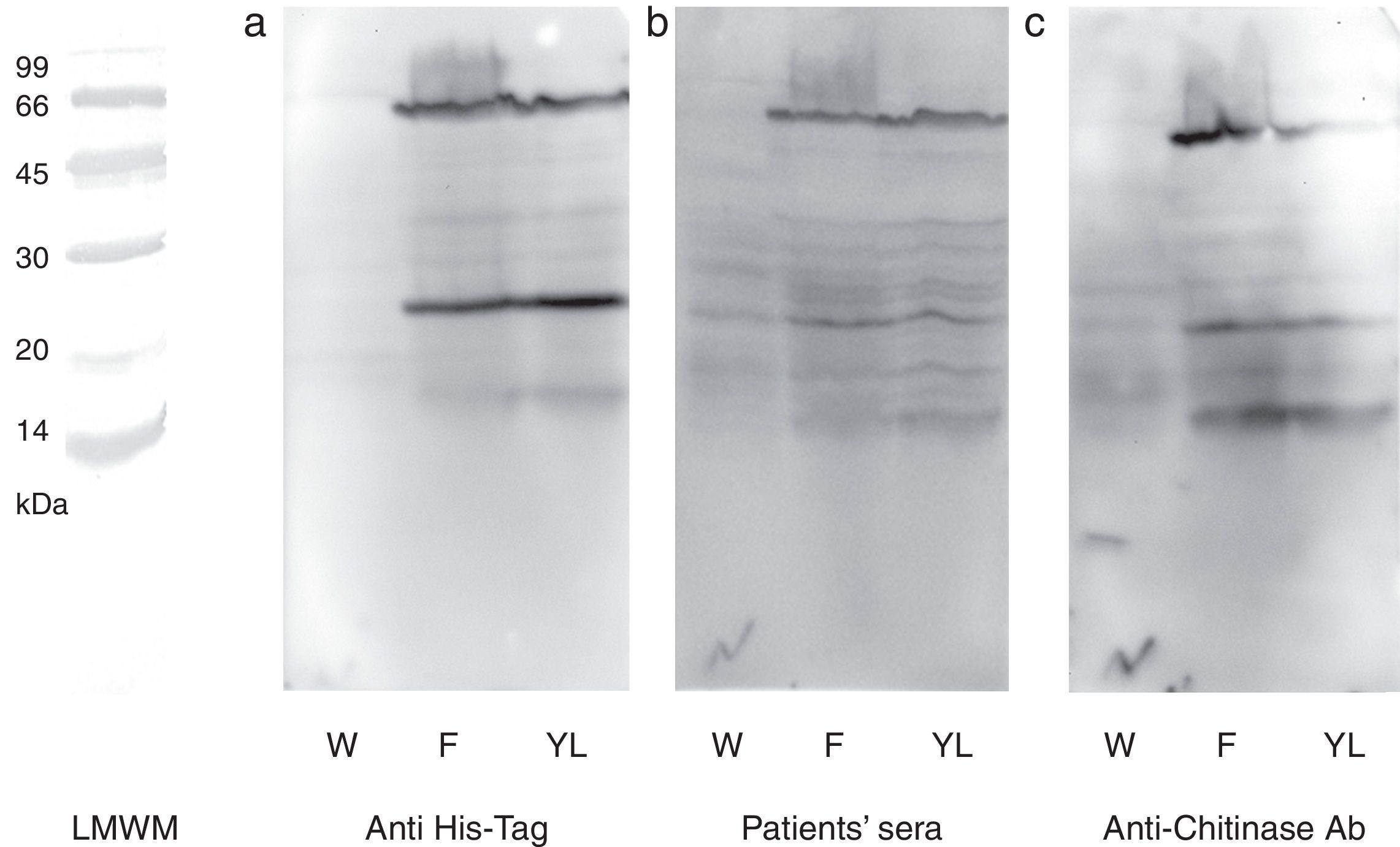
N-terminal 6His-tag containing recombinant protein was detected by commercial antibody. The anti-6His-tag antibody showed two distinct reactive bands at the mentioned spots (Fig. 3a). A pooled sera from grape class IV chitinase sensitive patients showed IgE reactivity with the produced proteins (Fig. 3b). Finally, monoclonal anti-chitinase antibodies reacted with the same bands. Moreover, a third faint, but broad, band was observed, approximately at 15kDa region in Western blotting with anti-chitinase antibody (Fig. 3c). This extra band is most probably an indicator of a degraded C-terminal part of the recombinant protein (a fragment without His-tag) which does not show reactivity with anti-6His-tag antibody. Mock-infected cell lines which were used as control, did not show significant reactions in any of the mentioned Western blotting assays.
 or wild type baculovirus infected Sf9 cells with (a) anti-6His-tag antibody, (b) grape allergic patients’ pooled sera and (c) anti-class IV chitinase antibody. Note that Sf9 cells were infected with wild type baculovirus (W) or chitinase expressing recombinant baculovirus (F and YL). The cells were grown in Grace")
Western blotting of grape class IV chitinase with different antibodies. Western blotting of the cell lysate from recombinant (coding for grape class IV chitinase) or wild type baculovirus infected Sf9 cells with (a) anti-6His-tag antibody, (b) grape allergic patients’ pooled sera and (c) anti-class IV chitinase antibody. Note that Sf9 cells were infected with wild type baculovirus (W) or chitinase expressing recombinant baculovirus (F and YL). The cells were grown in Grace's insect cell media containing 10% FCS (F) or Grace's media supplemented with 10% yeast lysate and lactalbumin hydrolysate (YL).
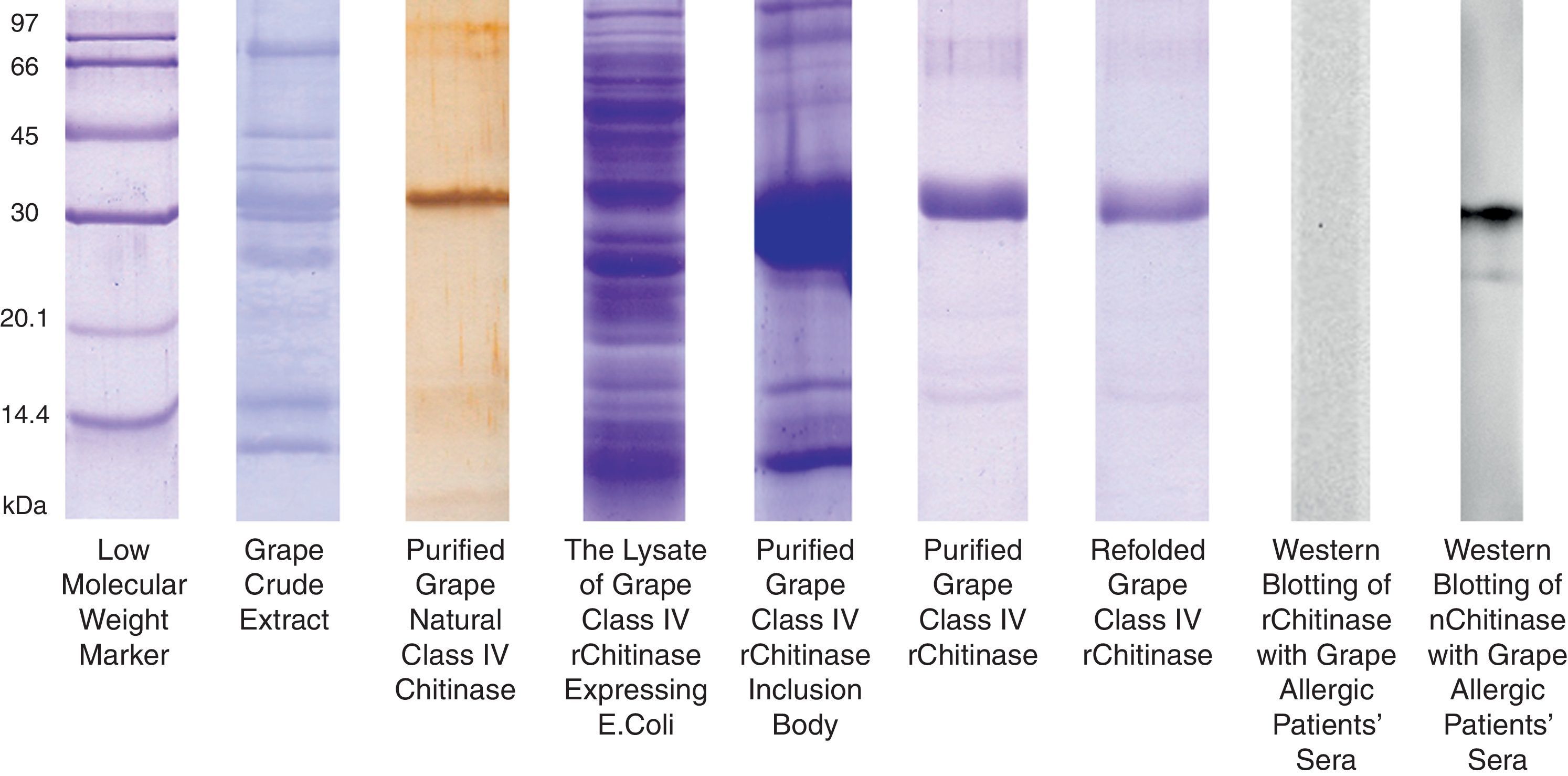
The grape chitinase sequence was more easily cloned and expressed in bacterial systems. As confirmed by DNA sequencing, the cloned fragment showed complete identity with the fragment that was inserted in baculoviral transfer vector at the same time. SDS–PAGE analysis of total bacterial lysate showed that an approximately 25kDa recombinant protein was highly expressed within inclusion bodies in both transformed bacterial hosts. In order to facilitate the protein refolding; the C-terminal 6His-tag fused recombinant protein was purified by metal affinity chromatography using the hybrid method. Although the refolded protein was soluble at low protein concentration, it did not show significant immunoreactivity with grape allergic patients’ sera (Fig. 4). Meanwhile, a similar amount of the natural form of the protein which had been previously fractionated by anion exchange chromatography21 showed significant IgE-reactivity with patients’ sera. However, both bacterially expressed proteins demonstrated specific reactivity with anti-6His-tag as well as monoclonal anti-chitinase antibodies, confirming their partial refolding.

SDS–PAGE analysis of natural and recombinant grape class IV chitinase and evaluation of their IgE reactivity. Grape class IV chitinase was cloned and expressed in bacterial host and the IgE-reactivity of the refolded protein was determined by Western blotting. Although the natural protein showed considerable reactivity with grape allergic patients’ pooled sera; the recombinant form which was expressed in E. coli Origami strain did not show significant IgE-reactivity.
In this study the Bac-to-Bac system was applied for the construction of a recombinant baculovirus which was later used for transfection and consequently expression of the recombinant allergen in Sf9 cells.
SDS–PAGE analysis of the cell lysates showed that the recombinant protein expression level was much lower in Sf9 cells, as compared to that of the bacterial host. Moreover, the protein was produced in comparable amounts of monomeric and dimmeric forms with apparent molecular weight of 25 and 50kDa. The molecular characteristic of 25kDa protein was consistent with the predicted molecular weight of previously reported grape class IV chitinases.24 Since at least one reduced cysteine remained after proper refolding; the dimmeric form could be the consequence of disulphide bridges between two distinct proteins. Although the expression level was low and the produced protein was expressed in monomeric and dimmeric forms, both of them showed IgE reactivity in a comparable intensity, as shown by Western blotting. This comparable immunoreactivity is also an indicator of proper refolding and dimmerisation of the protein.
The baculovirus expressed protein was immunocharacterised by three different Western blotting assays. Application of commercial anti-6His-tag antibodies revealed that both of the monomeric and dimmeric proteins contained 6His-tag and have been produced under the control of polyhedrin promoter by genetically modified baculovirus. Likewise, Western blotting with grape allergic patients’ sera (containing specific IgE to grape class IV chitinase) revealed that both of the expressed proteins reacted specifically with the patients’ serum IgE, proposing potential applicability of the expressed protein in diagnostic procedures. Moreover, specific immunoreactivity of the anti-class IV chitinase monoclonal antibodies with both expressed proteins confirmed that they belonged to the class IV chitinase family.34 However, the anti-chitinase Western blotting showed a weak reactive band approximately at 15kDa; indicative of a cross reactive protein in cellular crude extract. Since this immunoreactive band was not observed in mock-infected cells, and it was also absent in anti-6His-tag blotting; we assumed that it could be a proteolysed form of the recombinant protein.
Although the baculovirus encodes an active chitinase,35 there is no structural similarity between grape and baculovirus chitinases to interfere in diagnostic immunoassays. Baculovirus chitinase belongs to Glyco-18 superfamily (Accession No. NP_054156), while grape class IV chitinase is a member of Glycol-19, lysozyme-like superfamily (Accession No. AAB65777).
SDS–PAGE and Western blotting of the cell lysate showed that the apparent molecular weight of monomeric recombinant form (approximately 25kDa) was slightly lower than that of the purified natural form (approximately 28kDa), which could be due to variations in PTM such as the presence of potential N-glycosylation sites and other minor differences in mechanisms of PTM in plant and insect cells.
Baculoviruses have also been applied as biological pesticides. The constitutively produced wild form of baculovirus chitinase (AcMNPV ChiA) is partly responsible for killing and liquefaction of the baculovirus infected pests. Some studies have been performed to re-programme or increase the expression rate of baculoviral chitinases; leading to faster destroying of the plant pests.36 In the present study, the genetically constructed baculovirus produced a recombinant chitinase which might increase the chitinolytic effects of the baculovirus on infected pests, making it a useful tool to be applied as a new biopesticide in the future.
In addition to the baculovirus system, we employed a bacterial system to check if the prokaryotic systems are capable of producing an immunoreactive form of this protein. A rather similar set of primers were applied in both systems, so that identical DNA sequences were cloned as confirmed by DNA sequencing. Although those proteins could have equal primary protein structures, the evolutionary differences between prokaryotic and eukaryotic systems resulted in the production of recombinant proteins with different immunoreactivity.
The bacterial hosts only produced the protein as inclusion bodies. Optimisation of expression conditions, as well as the application of two different hosts did not result in expression of a soluble form of the protein. Solubilisation of the inclusion bodies in chaotropic solvents and consequently purification and refolding of the recombinant protein revealed that the produced molecule is not IgE reactive, while a purified natural form showed a strong IgE-reactivity with grape allergic patients’ pooled sera. Most probably, this occurred due to improper three-dimensional conformation of the recombinant protein corresponding to lower capability of the bacterial hosts for proper disulphide bond formation. In fact, this protein contains 15 cysteines that may form several random disulphide bonds when expressed in common bacterial hosts. Although, Origami strain contains thioredoxin reductase pathway components which may contribute to disulphide bond formations; it is not usually capable of correctly reforming randomly built disulphide bonds.
Taken together, it is believed that the baculovirus expression system may express plant or animal allergens in a nearly similar three-dimensional structure and comparable post translation modifications to their native forms.10 The proper folding of the expressed molecules may lead to the formation of conformational epitopes as seen on natural allergen, and consequently preserve the IgE-reactivity of the recombinant protein.37 This study confirmed that such a goal could be achieved for plant chitinases. Overall, plant PR proteins comprise a main group of plant allergens with a complicated structure which their immunoreactive forms rarely produced in bacterial systems. However, in most of those studies, production of the allergen in P. pastoris resulted in appropriate immunoreactivity. In the case of chitinases as a main family of PR proteins and as a major allergen of fruits, results were obtained which were similar and consistent with the findings of this study.38–40
ConclusionIn conclusion; in this study an IgE reactive grape class IV chitinase was expressed in insect cells using baculoviral system and in the case of high level expression it could be applied for immunodiagnostic or biotechnological purposes. However, the bacterial system could not produce an immunoreactive form of this protein.
Ethical disclosuresPatients’ data protectionThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Conflict of interestThe authors have no financial conflicts to disclose. All authors have read and approved the final version of the article.
This study was supported by Grant 88452 from the Research Administration Department of Mashhad University of Medical Sciences. This article is derived from the Ph.D. thesis of the first author (Thesis No. A-277).
