




Nonalcoholic liver disease (NAFLD) is a major emerging health burden that is a common cause of illness and death worldwide. NAFLD can progress into nonalcoholic steatohepatitis (NASH) which is a severe form of liver disease characterized by inflammation and fibrosis. Further progression leads to cirrhosis, which predisposes patients to hepatocellular carcinoma or liver failure. The mechanism of the progression from simple steatosis to NASH is unclear. However, there are theories and hypothesis which support the link between disruption of the bile acids homeostasis and the progression of this disorder. Previous studies have been demonstrated that alterations of these pathways can lead to dysregulation of energy balance and an increase of liver inflammation and fibrosis. In this review, we summarized the current knowledge of the interaction between BA and the process related to the development of NAFLD, besides, the potential targets for novel therapies.
Nonalcoholic fatty liver disease (NAFLD) is a major health burden and independent cause of cardiovascular disease. Some studies note that the worldwide prevalence of NAFLD is approximately 20%-30%1,2 with 2%-44% in Europeans and 15% in Asians.3,4 Given the lack of epidemi-ological data in the Americas, our group based on the prevalence of obesity to estimate the NAFLD prevalence in Mexico as 26% and 29% in the USA.5 Following these trends, population growth will increase the incidence of this disorder.
NAFLD is defined by the presence of steatosis, steato-hepatitis, and in some cases, the development of fibrosis and cirrhosis, including liver cancer.6 By contrast, the pathogenesis is multifactorial and not completely understood. Interestingly, Brunt, et al.7 proposed different key points in lipid accumulation, including weight gain, which is associated with an increase in adipose tissue and consequently the dysfunction and death of adipocytes. Dysfunctional adipocytes result in local inflammation and the upregulation of cytokines, which promote insulin resistance. In turn, this condition disables the ability of adi-pocytes to store fat, resulting in the release of high levels of free fatty acids into the circulation, which will available to be captured by various organs, such as the liver. This is made possible by fatty acid transport protein 5 (FATP5/ acylCoAsynthetase) and CD36, which is a surface receptor that facilitates uptake of these fatty acids.7 An increase of FATP5/acylCoA synthase in the liver stimulates the production of triglycerides, gluconeogenesis activation, hy-perglycemia, and an increase in the synthesis of compensatory insulin. However, the excess of fatty acids put an strain on hepatocyte mitochondria, which produces a dysfunction after the hepatocyte death. Hepatocyte death can be caused through two main pathways. The first is by endoplasmic reticulum stress, which leads to mito-chondrial dysfunction. This action is regulated by a cas-pase 2-mediated cleavage excision of BH3 death agonist in an interactive domain (BID). The second is through the activation of death receptors. The most common examples are the death ligands FAS and DR5, which eventually result in apoptosis and necroptosis of hepatocytes.8 In addition, it is well known that participation of the liver in total lipids storage, through a process called de novo lipogenesis (DNL), increases the risk to develop NAFLD.8-10
This review will focus on the role of BA which are considered the newly identified players in the complex pathogenesis and treatment of NAFLD.
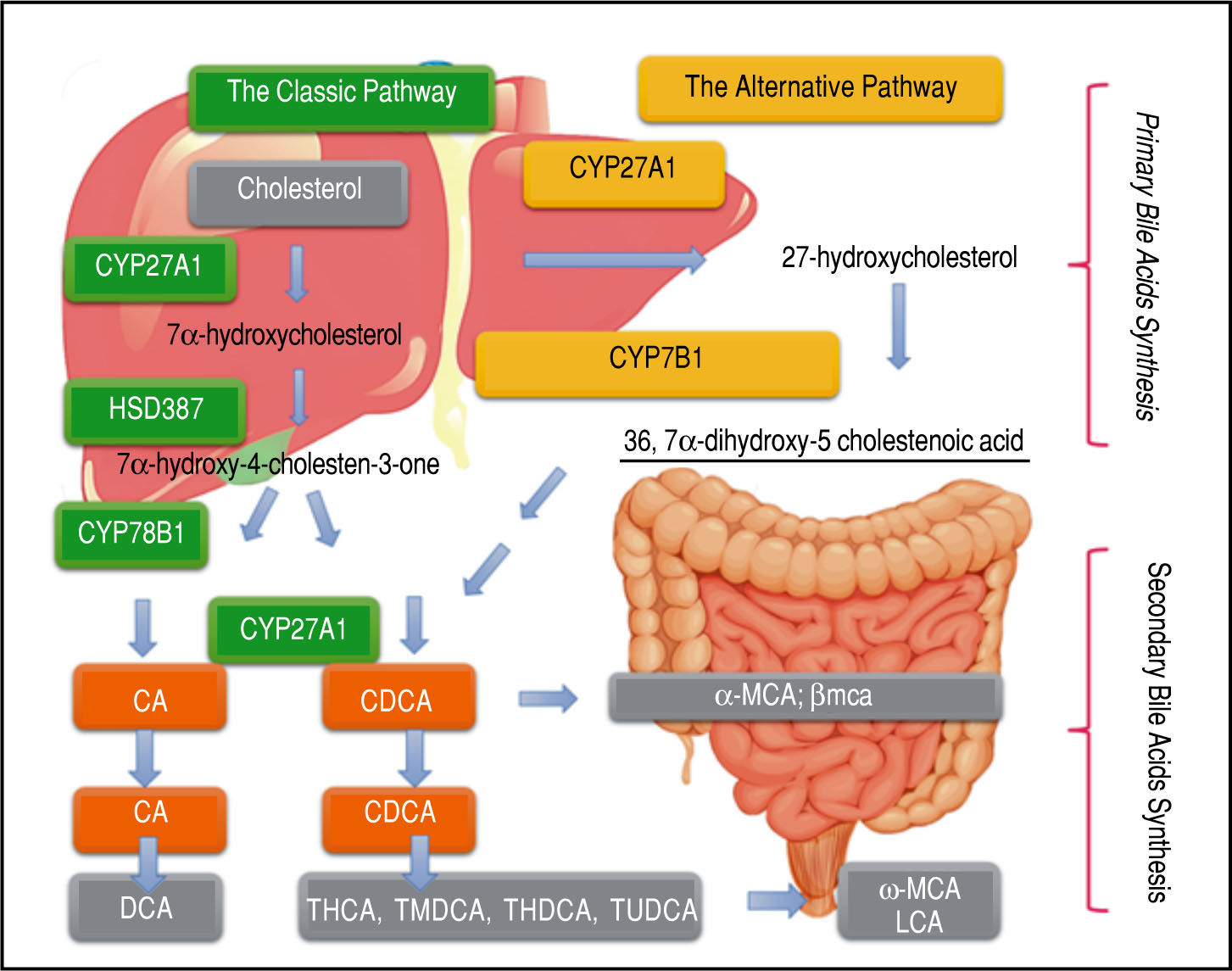
Light and Dark Sides of Bile Acids in NafldA major component of bile is bile acids (BA), which are amphipathic molecules synthesized in hepatocytes from cholesterol. The process of their synthesis has been known for several decades11 (Figure 1). The hydrophobic and hydrophilic regions of these amphipathic molecules can produce two great distinctive effects. The hydrophilic area plays a protective role for liver cells, while the hydro-phobic region can be cytotoxic and generate oxidative stress by inducing mitochondrial dysfunction and formation of reactive oxygen species, ultimately leading to apop-tosis or necrosis.12
, which is different from mi-crosomal cholesterol 7α-hydroxylase. Primary bile acids are metabolized by gut bacteria to form the secondary bile acids, DCA and LCA.")
Pathways and Synthesis: BA Cholesterol serves as the sole substrate and is converted to cholic acid via both classic and alternative pathways. Micro-somal cholesterol 7 -hydroxylase is the initial and rate-controlling enzyme for the classic pathway, whereas mitochondrial sterol 27-hydroxylase initiates the alternative pathway and might be rate limiting. The product, 27-hydroxycholesterol, is then 7α-hydroxylated by microsomal oxysterol 7α-hydroxylase (27-hydroxycholesterol-7α-hydroxylase), which is different from mi-crosomal cholesterol 7α-hydroxylase. Primary bile acids are metabolized by gut bacteria to form the secondary bile acids, DCA and LCA.
Other important functions of BA include the emulsifi-cation of dietary fats and intestinal absorption of lipids and lipophilic vitamins. BA are well known to play a critical role as regulators of hepatic lipid and glucose metabolism through farnesoid X receptor (FXR), vitamin D (NR111), and pregnane X (NR112), which are members of the nuclear receptor superfamily (NRS), and G protein-coupled receptor 5 (TGR-5) and sphingosine-1-phosphate 2, which are members of the G protein-coupled receptor su-perfamily.9,13 The ligands of NRS, such as peroxisome proliferator-activated receptors (PPARs) contribute to pathogenesis in metabolic diseases. PPARs oc (NR1C1), PPARs 8 (NR1C2), and PPARs y (NR1C3) are the three isotypes that are known. PPARs control the expression of a wide range of genes that maintain the metabolism of glucose, triglycerides (TG), and lipids, besides the synthesis, oxidation, storage, and export of BA.14 The disruption of these metabolic processes contributes to the pathogenesis of metabolic diseases, such as obesity, metabolic syndrome (MS), diabetes, NAFLD, and atherosclerosis.15
Current research has identified promising targets associated with BA Specifically, through their FXR and TGR-5 receptors, BA improve lipid and glucose homeostasis and inhibit the inflammatory response.9
Bile Acids and Fxr on Metabolism of Glucose and LipidsBA are important cell-signaling molecules that stimulate diverse pathways to regulate biological processes. The regulatory functions of BA are the result of modulation of intracellular ligand-activated nuclear receptor super-families (NRS), such as FXR and TGR-5.16-18
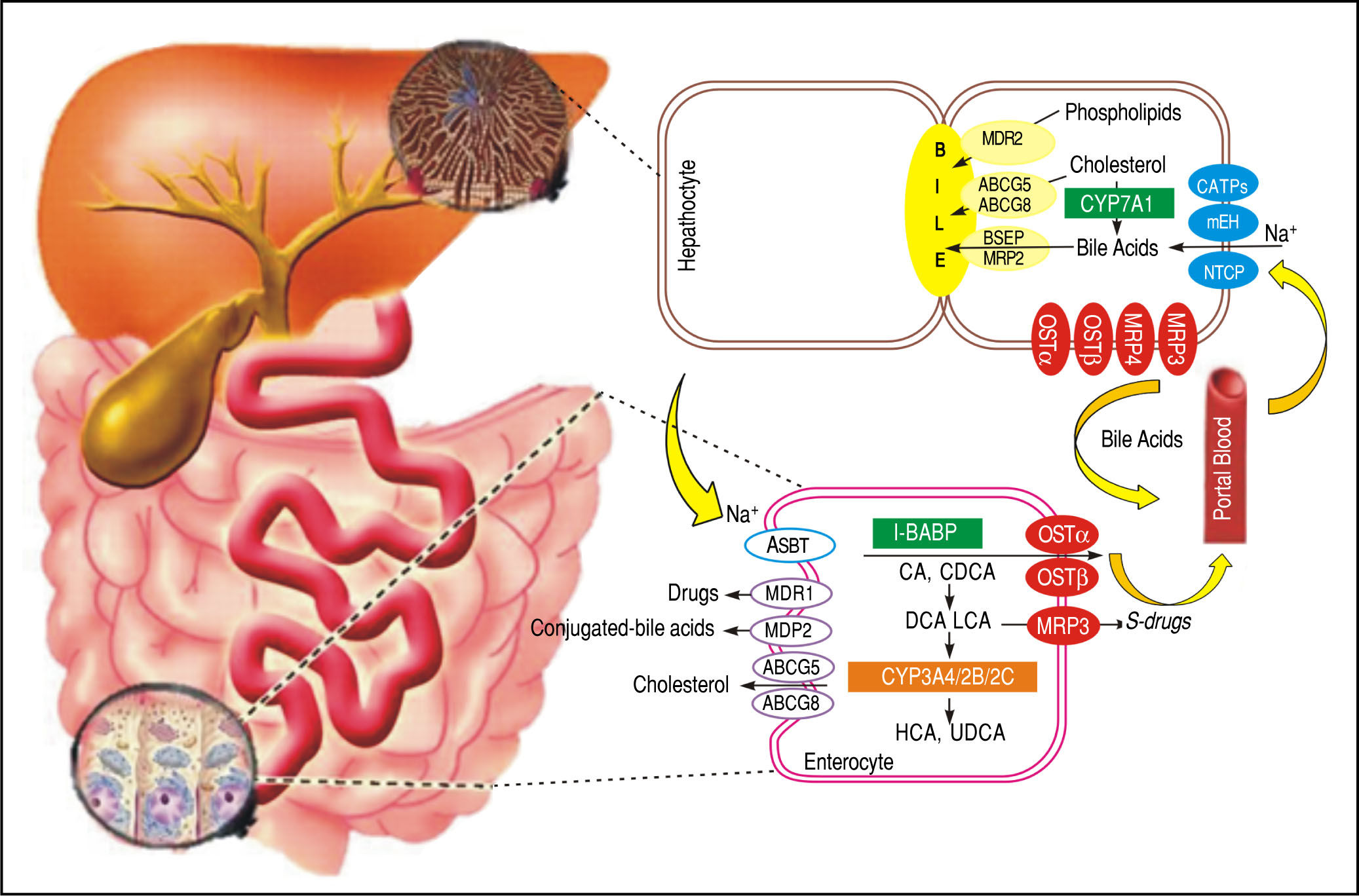
Additionally, FXR plays an important role in BA homeostasis controlling the metabolism of genes such as the nuclear receptor small heterodimer partner(SHP),19 Na+-taurocholate cotransporting polypeptide (NTCP), cholesterol 7α hydroxylase (CYP7A1), and the bile salt export pump (BSEP).20-22 The main functions of FXR can be summarized as follows. An increase in BA levels activates FXR, which suppresses their synthesis. This suppression occurs through induction of SHP, a nuclear receptor that binds to and interferes with the positive regulation of gene expression by other nuclear receptors, such as liver receptor homolog 1 (NR5A2) and liver X receptor (NR1H3).23,24 These nuclear receptors are involved in the control of genes that participate in BA synthesis and transport. This transport is conducted by NTCP, which transports BA from the circulation to the liver, and BSEP, which is directly activated by FXR and transports this molecule from hepatocytes to the gallbladder.25 Thus, when BA levels rise in the liver, CYP7A1, NTCP, and BA uptake from the blood is decreased, and BSEP and BA transport to the intestine is increased simultaneously (Figure 2).
. BA efflux is m ediated by the OST and OST heterodimers at the basolateral membrane. At the apical membrane of the enterocytes, ABCG5 and ABCG8 heterodimers transport cholesterol back into the intestinal lumen, a process that confines intestinal cholesterol absorption. CYP3A4, CYP2B, and CYP2C are implicated in the metabolism and detoxification of LCA in the intestine. MDR1 effluxes drugs and MRP2 effluxes conjugated BA in the apical membrane of intestine. At the sinusoida l membrane, the MRP3 output sulfur conjugated drugs for renal excretion.")
BA transporters in the hepatocytes and enterocytes the microsomal epoxide hydrolase and NTCP may be responsible for Na+-dependent uptake of conjugated BA at the basolateral membrane of the hepatocytes, whereas OATPs show substrate specificity for unconjugated BA. At the canalicular membrane of the hepatocytes, the BSEP performs a main role in biliary secretion of BA, while the MRP2 regulates secretion of organic substrates including glutathione, bilirubin, and BA. ABCG5 and ABCG8 heterodimers transport cholesterol into the bile, whereas MDR2 is responsible for biliary secretion of phospholipids. At the basolateral membrane of the hepatocytes, organic solute transporters OST and OSTβ heterodimers, MRP3, and MRP4 mediate the BA secretion into the circulation. With cholestasis, both basolateral BA efflux and renal BA excretion are increased. After BA are released from the gallbladder into the intestine, ileal BA uptake is regulated by the ASBT. Intracellular BAs are matched to the intestinal BA-binding protein (I-BABP). BA efflux is m ediated by the OST and OST heterodimers at the basolateral membrane. At the apical membrane of the enterocytes, ABCG5 and ABCG8 heterodimers transport cholesterol back into the intestinal lumen, a process that confines intestinal cholesterol absorption. CYP3A4, CYP2B, and CYP2C are implicated in the metabolism and detoxification of LCA in the intestine. MDR1 effluxes drugs and MRP2 effluxes conjugated BA in the apical membrane of intestine. At the sinusoida l membrane, the MRP3 output sulfur conjugated drugs for renal excretion.
By contrast, in ileum enterocytes, FXR activation, by BA agonists produced in the liver, improves the transport of BA from the gut lumen to the blood by inducing expression of apical sodium-BA transporters (ASBT, SLC10A2) and the organic solute and steroid transporter (OSTα, OSTβ) that convey BA from the enterocytes to the blood.26 Nevertheless, FXR has another pathway which controls the synthesis of BA in the liver. This is performed through the expression of the fibroblast growth factor 15 (FGF15) gene, designated FGF19 in humans, in enterocytes. FGF15/19 is transported to the liver and binds to and activates the hepatocyte plasma membrane receptor complex FGFR4β-Klotho, resulting in the suppression of CYP7A1 expression27 and then a decrease of BA synthesis.27,28
In addition, LXRα, a nuclear receptor of liver, is involved in NAFLD pathogenesis because it has been found to be disturbed in patients with NAFLD.29 This nuclear receptor has an important role in regulation of cholesterol metabolism and hepatic free acid biosynthesis.30 In this context, it has been observed that LXRα mRNA is increased, in rat models of NAFLD, under a fatty diet regi-men.31,32 Therefore, a positive correlation between the expression of LXRα and the degree of NAFLD was established because of a major presence of LXR expression that will cause more DNL, which ultimately results in more infiltration of fat into the liver.
Patients with nonalcoholic steatohepatitis (NASH) are well known to have raised levels of BA, both in liver tissue and plasma,33,34 suggesting a relationship between toxic levels of BA and the development of NASH. The understanding of the contribution of NRS and BA dysregulation for the pathogenesis and advancement of NAFLD is a principal issue in the Hepatology field.35
Patients with NASH may express different genes and proteins than patients with simple steatosis (SS). The expression of FXR, LXRcc, SHP, and NTCP in liver biopsy samples between patients with SS and NASH have been compared. Individuals with NASH possess a different mRNA and protein-expression profile of FXR, SHP, and NTCP versus patients with SS. An elevated mRNA expression of NRS (FXR and SHP) and NTCP in liver biopsies from patients with NASH was observed, whereas by contrast, the protein levels were decreased in the same samples. Otherwise, LXRcc gene expression and its protein level between the SS and NASH groups were not found to be significantly different.36 This illustrates that mRNA expression may not always be equivalent to protein level, a stark contrast to general assumption that protein levels must be correlated to the levels of their corresponding mRNAs. Many studies sustain that this general assumption may be incorrect in most cases.37-39 However, more studies are required to comprehend molecular mechanisms of BA regulation more fully. In turn, this will allow us to understand the progression of NAFLD more thoroughly.40
Bile Acids and Tgr-5TGR-5 plays a regulatory role in the glucose homeostasis through the enteroendocrine cells, which are able to induce glucagon-like peptide 1 (GLP1), a potent anti-inflammatory molecule capable of blocking the action of cytokines and infiltrated macrophages by nuclear translocation of nuclear factor kB (NF-kB).41 Induction of GLP1 by TGR-5 in obese mice leads to improvement in liver and pancreatic function and consequently glucose tolerance.
Resistance to weight gain and hepatic steatosis, preservation of liver and pancreatic function, maintenance of glucose homeostasis and insulin sensitivity are beneficial metabolic effects produced by TGR-5 activation. These effects result from improved mitochondrial function in muscle and enteroendocrine cells, leading to an increase in energy expenditure and incretin secretion.42,43
Body Mass Index and NafldObesity, defined as a body mass index (BMI) 30, is the principal risk factor to develop NAFLD and may take the first place in Latin American populations. A Mexican study estimated that there will be more obese people than overweight people by 2,050, resulting in widespread dysli-pidemia and insulin resistance.44 Ninety percent of obese individuals will have associated with MS.45 The NAFLD prevalence of 69% among patients with type 2 diabetes (T2D) has been estimated by Leite, et al. study.46 Many alterations in BA levels, which are increased in obesity, have been observed, consequently it has suggested that these alterations affect the energy metabolism.47
Previous studies48-50 in patients with insulin resistance and T2D concluded that plasma levels of BA are elevated. The cause of this alteration is unknown, but some possibilities have been proposed. First, the hydroxylation of BA has a preference for the 12α position in patients with insulin resistance, resulting in relatively greater synthesis. Second, insulin reduces serum levels of BA in healthy patients; in obesity, this effect is inhibited. Third, the levels of BA during the meal are attenuated with respect to fasting levels. Finally, the expression of BA transporters in the liver is negatively associated with BMI.
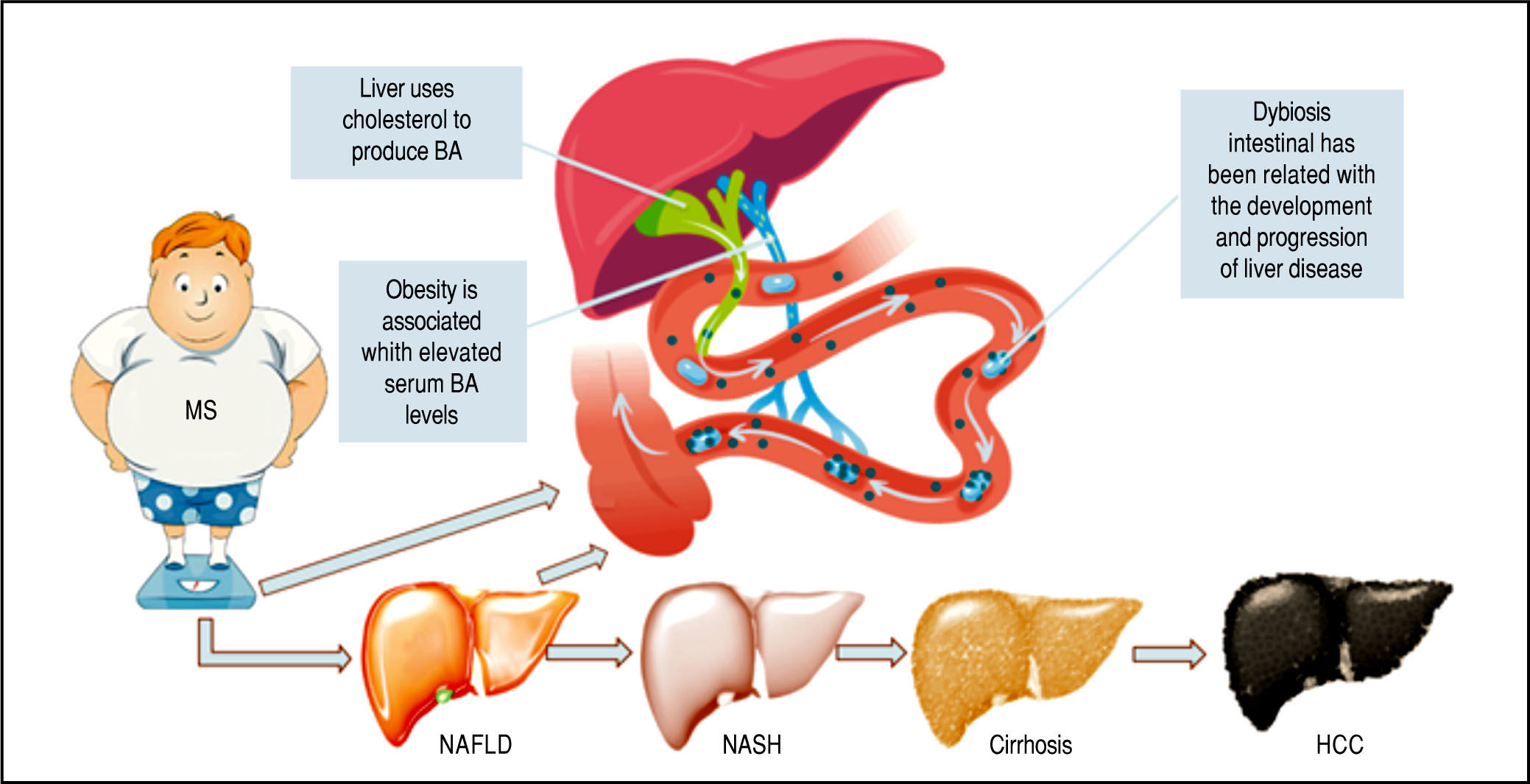
The Role of Microbiota in Ba MetabolismNAFLD is a hepatic expression of MS (Figure 3), being frequently related to insulin resistance, dyslipidemia, and obesity.51

Metabolic Syndrome: Obesity as the critical risk factor for Nonalcoholic Liver Disease MS is one of the main risk fac tor for NAFLD. This illustration shows the different stages of the disease and shows a schematic representation of the action of BA as signaling molecules in NAFLD.
A role for gut microbiota (GM) in insulin resistance, obesity, and associated metabolic disorders has been demonstrated, increasing the interest in GM’s relationship with NAFLD pathogenesis.52-55 Therefore, GM have appeared as a potential factor involved in NAFLD.
Human GM include 10-100 trillion microorganisms56 composed of bacteria, archaea, virus, and fungi. The four main phyla of bacteria are: Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria, which represent more than 95% of GM.57-59 The microbiome refers to the collective genome of the GM and contains many important genes for glycan and amino-acid metabolism, xenobiotic metabolism, methanogenesis, and biosynthesis of vita-mins.58 The GM regulates body fat gain and insulin resistance, so it seems to play an essential role in NAFLD through different pathways, including increasing energy harvest from diet, expression changes of genes involved in the DNL, regulation of choline metabolism, ethanol production, inflammasomes, and innate immunity and in-flammation.60
A disruption of the normal GM, dysbiosis, is linked to the pathogenesis of human liver disease. Early evidence of NAFLD associating gut dysbiosis with liver injury came from descriptive human studies of small intestinal bacterial overgrowth diagnosed by D-xylose and lactulose breath testing61,62 in an advanced stage of NAFLD. This early evidence coincides with current data that support the role of the microbiome in human diseases, such as obesity and its related disorders. Experiments in mice provide evidence that phenotypes can be altered by transfer of GM from obese animals to lean littermates.
GM has an important function in signaling pathways and immune responses. Therefore, it plays a central role in the development and progression of NAFLD. For this reason, an understanding of GM may provide novel therapeutic targets for improving NAFLD treatment.63
Novel Pharmacological Targets for the Treatment of NafldAccording to the natural history of the disease, more aggressive treatment is recommended for NASH than for NAFLD. In this section, we describe some of the 187 studies being conducted with the aim of evaluating the efficacy and safety of NASH treatments, of which 79 are completed and 51 are still recruiting at the time of writing64
AramcholIn 2014, an Israeli research group developed a drug called aramchol. This drug inhibits stearoyl-CoA desaturase 1 (SCD1), which produces a decrease of fatty acids synthesis and increase in P-oxidation. Consequently, there is a decline in the storage of TG and esters of fatty acids in the liver. Safa-di and his group evaluated the effect of aramchol using spectroscopy identifying a dose-dependent reduction in levels of hepatic steatosis from 100 to 300 mg vs. placebo.65
ElafibranorElafibranor is an agonist of PPARa/5. PPARa is a lig-and-activated nuclear receptor (NR1C) expressed in the liver that adapts the rates of fatty acid catabolism and lipo-genesis in response to feeding.66 Moreover, it controls the lipid flux in the liver by modulating fatty acid transport and (3-oxidation, while improving plasma lipids by decreasing TG and increasing high-density lipoprotein (HDL) cholesterol. By contrast, PPAR8 improves glucose homeostasis by upgrading insulin sensitivity and inhibiting the hepatic glucose output. This is the mechanism of elafibranor (a dual PPARcc/8 agonist), which reduces the steatosis, fibrosis, and inflammation in patients with NAFLD as confirmed by Ratziu, et al.68 after assessing its safety and efficacy. Interestingly, Ratziu, et al. showed that using the highest dose (120 mg) of elafibranor did not aggravate the liver fibrosis, but it could reverse NASH (considering “worsening” as any NAFLD stage that implicates fibrosis). The main adverse events identified in this phase 2 trial were nausea (10%), headache (7%-87%), diarrhea (5%-62%), and fatigue (5%-62%). GENFIT (Biopharmaceutical Company) is conducting a phase 3, multicenter, randomized, double blind, placebo-controlled trial to evaluate the efficacy and safety of this drug in a bigger population than the previous phase trials.
FXR agonistsFXR, as we mentioned before, are nuclear hormone receptors expressed in several tissues (including liver, bowel, and kidney) and play a determining role in carbohydrate and lipid metabolism, such as regulation of insulin sensitivity.69
Many preclinical studies have assessed the FXR agonists role in the development of NAFLD and their results have shown that they cause an improvement of hyperlipi-demia, enhanced glucose tolerance, and insulin sensitivity. These results have also been compared with various other diseases of humans, mostly in patients with NAFLD or primary biliary cirrhosis (PBC) (70) (Table 1). Obeti-cholic acid (OCA), a derivative of CDCA and a first-in-class selective FXR agonist, has shown two outcomes. The first included important increases in insulin sensitivity, and reduced markers of liver inflammation and fibrosis in patients with NAFLD and with T2D.71 The second was changes in body weight, liver enzymes, serum lipids, FGF19, 7cc-hydroxy-4-cholesten-3-one (C4, a cholesterol metabolite formed by the rate-limiting enzyme CYP7A1), endogenous BA, and serological measures of liver fibrosis and apoptosis. Patients in the group given an OCA dose of 50 mg experienced a significant average weight loss of 2% (P = 0.008) compared with patients given a placebo.72 In concordance with the mechanism of OCA, the FXR ago-nism resulted in an increase of FGF19 and a reduction of endogenous BA and C4 production.
Clinical trials and ongoing studies of FXR receptor agonists in NAFLD.
| Agent | Indication | Results and Comments |
|---|---|---|
| OCA (25 and 50 mg daily) for 6 weeks | NAFLD and patients with type II diabetes | OCA was well tolerated, increased insulin sensitivity and reduced markers of liver inflammation |
| OCA (25 mg daily) for 72 weeks (42) | Non-cirrhotic NASH | OCA improved histology in 45% of patients, compared with 21% in the placebo. Pruritus occurred in 23% of patients in the OCA arm |
| CA (15 mg/kg/day for 6 months) | NAFLD in patients with lipodystrophy | CA did not reduce hepatic TG or biochemistries compared to placebo |
| px-104 | NAFLD | CA was well tolerated. Study ongoing. |
Recently, 6a-ethyl-3a,7a-dihydroxy-24-nor-5(3-cho-lan-23-sulfate (INT-767), a potent dual FXR and TGR-5 agonist, has demonstrated potential for NAFLD treatment in obese diabetic mice. McMahan, et al. observed that treating diabetic (db/db) obese mice with INT-767 decreased hepatic steatosis, and reduced proinflammatory cytokine expression, directed monocytes and macrophag-es toward an anti-inflammatory M2 phenotype. They suggested that the modulation of FXR and TGR-5 may be useful in NAFLD treatment by improving insulin secretion and sensitivity and controlling glucose, lipids, and BA homeostasis.73
In addition, the treatment of obese mice with INT-767 had shown significant reductions of total plasma cholesterol and TG levels.61 By contrast, in a MDR2-/-mouse model of chronic cholangiopathy, INT-767 was able to ameliorate hepatic injury by decreasing biliary BA output and promoting HCO-3-rich bile secretion.74
According to this, INT-767 could provide new opportunities for the treatment of metabolic diseases, such as T2D and obesity, as well as chronic liver diseases.
Inhibitors of bile acid absorptionBile acid sequestrants (BAs), such as colesevelam and cholestyramine, reduce plasma low-density lipoprotein levels and improve glycemic control. They act by promoting cholesterol catabolism through BA biosynthetic pathways and improving hepatic insulin sensitivity. Thus, BA sequestration can be fruitful for NAFLD treatment.75,76
However, current experimental and clinical data do not support this argument. Le, et al. specifically designed a study to evaluate the efficiency of colesevelam in decreasing liver fat in patients with biopsy-proven NASH obtaining a small increase in liver fat detectable through magnetic resonance imaging and spectroscopy.77
Furthermore, Solis, et al. showed that BAs neither produce any positive effects on liver steatosis in ob/ob mice nor histology nor hepatic triglyceride content was influenced by cholestyramine.
BAs might not be as helpful for the treatment of NAFLD as once was assumed. Therefore, it is important to reconsider the proposed role of BA in the treatment of diabetic patients, considering that they are a population with a high risk of liver complications such as NAFLD and its potentially progressive form NASH.78
ConclusionDespite its complex pathogenesis and our partial understanding, we conclude that BA play an important role in NAFLD, mostly inducing cytotoxicity that leads to apop-tosis or necrosis of hepatocytes. FXR agonists have shown promising results for maintaining glucose and lipid home-ostasis. However, it is still necessary to continue assessing and clarifying their safety, efficacy, and related adverse events.
Abbreviations- •
ABCG5: ATP-binding cassette subfamily G member 5.
- •
ABCG8: ATP-binding cassette subfamily G member 8.
- •
ASBT: apical sodium-dependent bile acid transporter.
- •
BA: bile acids.
- •
BAs: bile acid sequestrants.
- •
BH3: bcl-2 homology 3.
- •
BID: death agonist in an interactive domain.
- •
BMI: body mass index.
- •
BSEP: bile salt export pump.
- •
C4: complement component 4.
- •
CA: cholic acid.
- •
CD36: cluster of differentiation 36 or platelet glyco-protein 4.
- •
CDCA: chenodeoxycholic acid.
- •
CYP3A4: cytochrome P450 3A4.
- •
CYP2B: cytochrome P450 2B.
- •
CYP2C: cytochrome P450 2C.
- •
CYP7A1: cholesterol 7 hydroxylase.
- •
DB/DB: diabetic mice/nonfunctional leptin receptors.
- •
DCA: deoxycholic acid.
- •
DR5: death receptor 5.
- •
DNL:de novo lipogenesis
- •
FAS/TNFRS6: tumor necrosis factor receptor super-family member 6.
- •
FATP5 /acyl coA: fatty acid transport protein 5.
- •
FGF15: fibroblast growth factor 15.
- •
FGF19: fibroblast growth factor 19.
- •
FGFR4-β Klotho: fibroblast growth factor receptor 4-beta Klotho.
- •
FXR: farnesoid X receptor.
- •
GLP1: glucagon-like peptide 1.
- •
GM: gut microbiota.
- •
HDL: high-density lipoprotein.
- •
I-BABP: ileal bile acid-binding protein.
- •
INT 767: 6α-ethyl-3,7α-dihydroxy-24-nor-5β-cholan-23-sulfate.
- •
LXRα/NR1H3: liver X receptor α.
- •
LCA: lithocholic acid.
- •
M2: muscarinic acetylcholine receptor.
- •
MDR1: multidrug resistance gene associated protein 1.
- •
MDR2: multidrug resistance gene associated protein 2.
- •
MRP2: multidrug resistance-associated protein 2.
- •
MRP3: multidrug resistance-associated protein 3.
- •
MRP4: multidrug resistance-associated protein 4.
- •
mRNA: messenger RNA.
- •
MS: metabolic syndrome.
- •
NAFLD: nonalcoholic fatty liver disease.
- •
NASH: nonalcoholic steatohepatitis.
- •
NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells.
- •
NR1C: activated nuclear receptor.
- •
NR1H3: nuclear receptor subfamily 1, group H, member 3.
- •
NR5A2: nuclear receptor subfamily 5 group A member 2.
- •
NR111: vitamin D receptor.
- •
NR112: pregnane X receptor
- •
NRS: nuclear receptor superfamily.
- •
NTCP: Na+-taurocholate cotransport peptide.
- •
OATPS: organic anion transporter.
- •
OCA: obeticholic acid.
- •
OST: organic solute transporter.
- •
OSTα: organic solute transporterα.
- •
OSTβ: organic solute transporter β.
- •
PBC: primary biliary cirrhosis.
- •
PPARs: peroxisome proliferator-activated receptors α.
- •
PPARα/NR1C1: peroxisome proliferator-activated receptor α.
- •
PPARδ/NR1C2: peroxisome proliferator-activated receptor δ.
- •
PPARγ/NR1C3: peroxisome proliferator-activated receptor γ.
- •
SCD1: stearoyl-CoA desaturase 1.
- •
SHP: small heterodimer partner.
- •
SLC10A2: solute carrier family 10 member 2.
- •
SS: steatosis simple.
- •
T2D: type 2 diabetes.
- •
TG: triglycerides.
- •
TGR-5: G protein-coupled receptor 5.



