



Cirrhosis of the liver is by far the most common cause of portal hypertension in the western world. Portal hypertension is a frequent clinical syndrome, defined by a pathological increase in the portal venous pressure. When the portal pressure gradient (the difference between pressures in the portal and the inferior vena cava veins: normal value below 6 mmHg) increases above 10-12 mmHg, complications of portal hypertension can occur. Increased resistance to portal blood flow, the primary factor in the pathophysiology of portal hypertension, is in great part due to morphological changes occurring in chronic liver diseases. However, more recently a graded and reversible contraction of different elements of the porto-hepatic bed have been shown to play a role modulating intrahepatic vascular resistance which provides a rationale for the intention to reduce intrahepatic resistance and portal pressure by means of pharmacological agents. The subsequent increase in portal blood flow, as a result of the arteriolar vasodilatation of the splanchnic organs, plays a contributory role maintaining and aggravating the portal hypertensive syndrome. This splanchnic arteriolar vasodilatation is a multifactorial phenomenon, which may involve neurogenic, humoral and local mechanisms.
Cirrhosis of the liver is by far the most common cause of portal hypertension in the western world. Portal hypertension is a frequent clinical syndrome, defined by a pathological increase in the portal venous pressure. When the portal pressure gradient (the difference between pressures in the portal and the inferior vena cava veins: normal value below 6 mmHg) increases above 1012 mmHg, complications of portal hypertension can occur. These complications represent the first cause of death and the main indication for liver transplantation in patients with cirrhosis.
Pathophysiology of portal hypertensionThe portal pressure gradient is determined by the product of portal blood flow and the vascular resistance that opposes that flow. Ohm’s law defines this relationship in the equation:
ΔP = Q x R
in which ΔP is the portal pressure gradient, Q is the flow within the portal venous system, and R is the vascular resistance of the portal venous system, which represents the sum of the resistance of the portal vein, the hepatic vascular bed, and of the portosystemic collaterals.
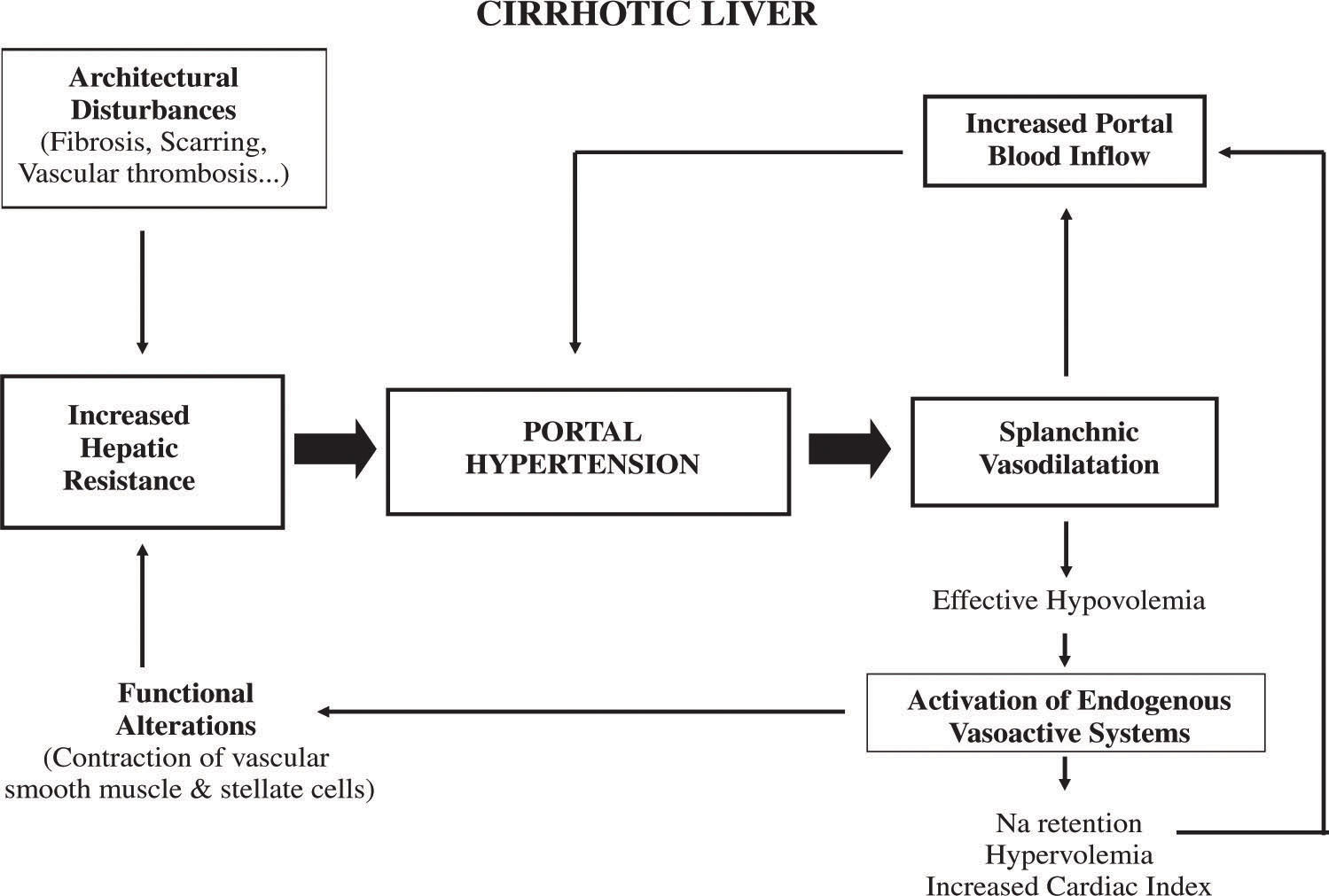
It follows that portal pressure may be increased by an increase in portal blood flow, an increase in vascular resistance, or a combination of both.1,2 However, it is well established that in cirrhosis, the primary factor leading to portal hypertension is an increased resistance to portal blood flow. Later on, an increase in portal venous inflow will help to maintain and aggravate portal hypertension (Figure 1).
Increased vascular resistance to portal blood flow
Increased resistance to portal blood flow may occur at any site within the portal venous system.
Although much of the increase in intrahepatic resistance is the mechanical consequence of the architectural disturbances caused by the cirrhotic process, it is clear that on top of these alterations there is an active contraction of several elements in the liver that further contribute to increase resistance. It has been claimed that this dynamic and reversible component may represent up to 40% of the total increased intrahepatic vascular resistance in cirrhosis. Contractile elements influencing the hepatic vascular bed can be located at sinusoidal as well as extrasinusoidal levels and include vascular smooth muscle cells of the intrahepatic vasculature (i.e. small portal venules in portal areas),3 activated hepatic stellate cells (HSCs) (pericyte cells located in the perisinusoidal space of Disse with extensions that wrap around the sinusoids and reduce its caliber after contraction)4,5 and hepatic myofibroblasts that may compress the regenerating nodules or venous shunts within the fibrous septa.
An increased production of vasoconstrictors and an exaggerated response of the hepatic vascular bed to them, as well as an insufficient release of vasodilators together with an insufficient response to vasodilators of the hepatic vascular bed are the mechanisms that have been implicated in the pathogenesis of the dynamic component of the increased intrahepatic resistance of the cirrhotic liver.
Increased production/exaggerated response of the hepatic vascular bed to vasoconstrictorsDifferent vasoconstrictive factors, that are detailed below, have been involved in the regulation of hepatic vascular tone in cirrhotic livers.6
Adrenergic agonistsThe alpha-adrenergic agonist norepinephrine, that is usually elevated in decompensated cirrhosis, has been shown to increase intrahepatic vascular resistance.7,8 This increase in resistance is completely blunted by the administration of a-adrenergic antagonists, such as prazosin. This agent by itself markedly reduces hepatic resistance and portal pressure in patients with cirrhosis. On the other hand, the administration of α-adrenergic agonists, such as isoproterenol, reduces intrahepatic vascular resistance in perfused cirrhotic liver. These data suggest that adrenergic receptors may be involved in the regulation of intrahepatic resistance in cirrhosis, and that β-adrenergic receptor blockers may decrease portal pressure in cirrhosis. In addition, the hepatic vascular bed of cirrhotic livers exhibits an exaggerated response to the β-adrenergic agonist methoxamine. It has been shown that the coupling of different agonists to its membrane G-coupled receptors promote the release of arachidonic acid from the plasma membrane facilitating its metabolization to different vasoactive-derived metabolites, including prostaglandins (PGs), thromboxanes (TXs), and leukotrienes.9,10
ThromboxaneCyclooxygenase (COX) is the key enzyme in the bio-synthetic pathway leading to PGs and TX from arachi-donic acid.11 COX-1 is constitutively expressed but it can also be stimulated by factors similar to those that stimulate the constitutive isoform of nitric oxide (NO) synthase (eNOs).12,13 COX-2 can also be constitutively expressed in some tissues including the liver14,15 and the mesenteric vascular bed.16 COX-2 is an inducible isoform of cyclooxygenase that, similarly to the inducible isoform of NO synthase, is usually expressed or over-expressed after stimulation with proinflammatory agents.17
The hyperresponse of the hepatic vasculature of cirrhotic livers to the vasoconstrictor methoxamine is associated with an overproduction by the isoenzyme COX-1 of thromboxane A2 (TXA2). This hyperresponse is completely corrected by pretreating the livers either with non-selective COX blockers, with COX-1 selective blockers or with TXA2 antagonists. Thus, an increased production of TXA2 markedly enhances the vasoconstrictive response of the cirrhotic hepatic vascular bed to methoxamine.14 Whether this effect is also shared by other vasoconstrictors has not been investigated so far.
Cystenyl leukotrienesCysteinyl leukotrienes (CT) are a group of highly potent vasoactive substances derived from the oxygenation and dehydration of arachidonic acid by 5-lipoxygenase18,19 that increases intrahepatic vascular resistance in normal and cirrhotic rat livers. However, this response is significantly greater in cirrhotic livers that in addition also have an increased expression of the 5-lipoxygenase mRNA and an increased production of CT. 5-lipoxygenase inhibition produces a marked reduction in portal pressure in cirrhotic livers, which suggests that 5-lipoxygenase—derived eicosanoids also contribute to the increased hepatic vascular resistance in cirrhosis.20
EndothelinsEndothelins are a family of homologous 21-amino acid vasoactive peptides (ET-1, ET-2, and ET-3) that also modulate hepatic vascular tone in cirrhosis.6,21 The biologic properties of endothelins are mediated essentially by two major endothelin receptors, endothelin A (ET-A) and endothelin B (ET-B) receptors. The ET-A receptor shows a high affinity for ET-1, but not for ET-3, and mediates constriction; the ET-B receptor has equal affinity for ET-1 and ET-3. Activation of ET-B receptors located on the vascular smooth-muscle cells promotes vasoconstriction, whereas activation of ET-B receptors located on endothelial cells promotes vasodilation, which is mediated by enhanced NO and prosta-cyclin production by the endothelial cell.
Patients with liver cirrhosis have increased circulating plasma levels of ET-1 and ET-3.22 ET-1 increases portal perfusion pressure by increasing intrahepatic resistance in normal and cirrhotic livers. While some experimental studies reported a slight reduction of portal pressure in cirrhotic animals after the administration of endothelin antagonists,23,24 this was not confirmed by other studies,25 so the role of endothelins increasing the vascular tone in cirrhosis remains unsettled.
Angiotensin IIAngiotensin II (A-II) is a powerful vasoconstrictor that increases hepatic resistance. Increased A-II is the result of the activation of the renin-angiotensin system (RAS), which is commonly observed in patients with cirrhosis. Activation of RAS may also be detrimental for portal hypertension as a result of increased liver fibrogenesis,26 which may worsen the evolution of cirrhosis. All the above suggested that preventing the activation of RAS may have beneficial effects in decreasing portal pressure in cirrhosis. However, current evidence shows that, although A-II blockade may reduce portal pressure, it causes systemic hypotension what reduces its potential as a therapeutic strategy for portal hypertension.27
EndocannabinoidsEndogenous cannabinoids (or endocannabinoids) is a collective term describing a novel class of endogenous lipid ligands, including anandamide (arachidonyl ethanolamide). Anandamide has been reported to induce both vasodilatation and vasoconstriction depending on its concentration as well as the vascular territory being examined. Recent studies suggest that anandamide might be involved in the increased resistance of cirrhotic livers by promoting an increase production of TXA2.28
Endothelial dysfunction of cirrhotic liversIn normal conditions, the endothelium is able to generate vasodilator stimuli in response to increases in blood volume, blood pressure or vasoconstrictor agents, in an attempt to prevent or attenuate the concomitant increase in pressure. In several pathological conditions there is an impairment in this endothelium-dependent vasodilatation, which has been named as «endothelial dysfunction».29,30 Endothelial dysfunction is considered one of the main pathological mechanisms involved in the increased vascular tone observed in several vascular disorders such as arterial hypertension,31 diabetes32 and hypercholesterolemia,33 and has been attributed to a diminished NO bioavailability29,30 and to an increased production of endothelial derived contracting factors (EDCFs), such as PGH2/TXA2,34 endothelin35 or anion superoxide.36 The hepatic vascular bed of cirrhotic livers also exhibits endothelial dysfunction.37 Indeed, contrary to what happens in normal livers, the cirrhotic liver can not accommodate the increased portal blood flow caused by the postprandial hyperemia, which determines an abrupt postprandial increase in portal pressure.38 In addition, in experimental models of cirrhosis, endothelial dysfunction has been further characterized by showing that the cirrhotic liver exhibits an impaired response to the endothelium-dependent vasodilator acetylcholine.15,37 Endothelial dysfunction in cirrhosis has been attributed to reduced NO bioavailability and to increased vasoconstrictor COX-1 derived prostanoids.
Reduced NO bioavailability within the cirrhotic liverNO is the natural ligand for soluble guanylate cyclase and is responsible for an increase in cyclic guanosine monophosphate (cGMP), the final agent responsible for the relaxation of the vascular wall through the extrusion of cytosolic Ca2+. Endothelial NO synthase (eNOS) is responsible for most of the vascular NO produced in a reaction where L-arginine is oxidized to L-citrulline and NO.39 In cirrhotic liver, there is a reduced NO bioavailability that plays a major role increasing intrahepatic vascular resistance and thereby worsening portal hypertension. In accordance with this concept, the administration of nitrates (exogenous donors of NO) have been shown to decrease portal pressure. In addition, enhancement of the expression of NO synthase in liver cells, through the portal injection of adenovirus coupled with the gene encoding NO synthase, significantly reduces portal pressure.40 More recently, strategies aimed at increasing NO release by enhancing intrahepatic eNOS activity, based on constitutively active Akt gene transfer41 or by simvastatin administration42 have opened new perspectives with potential therapeutic implications.
Decreased NO production occurs despite a normal expression of eNOS mRNA and normal levels of eNOS protein37,43 and has been attributed, at least in part, to reduced eNOS activity caused by several posttranslational alterations in the regulation of the enzyme such as increased caveolin expression, or a defect of the essential cofactor of eNOS, tetrahydrobioterin.44,45
Like in other vascular disorders characterized by the presence of endothelial dysfunction, oxidative stress has been implicated in the increased vascular tone that exhibit cirrhotic livers.46 It is well known that superoxide is capable of reacting with NO-, leading to peroxynitrite (ONOO-) formation,47,48 with an ongoing decrease in NO bioavailability.49-53 Reactive oxygen species (ROS) can also affect NO biology by its capacity to oxidize, and therefore inactivate, the NO synthase cofactor tetrahydro-biopterin (BH4),31 leading to a situation that has been called eNOS uncoupling,54-56 in which the NO synthases are incapable of transferring electrons to L-arginine and start using oxygen as a substrate leading to O2.- formation instead of NO. It has been suggested that because of that antioxidant therapy may contribute to correct this abnormality.57
Increased production of vasoconstrictor prostanoidsEndothelial dysfunction was also shown to be associated with an increased production of TXA2 and completely prevented by selective COX-1 blockers and by TXA2 antagonists. These results suggest that an increased production of a COX-1 derived vasoconstrictor prostanoid probably TXA2, is, at least in part, responsible for endothelial dysfunction.15 All these findings suggest that in cirrhotic livers there is an over-activation of the COX-1 pathway with an increased production of their vasoconstrictor-derived compounds.
Hydrogen sulphyde (H2S)H2S is produced endogenously from desulphydration of cysteine by three different enzymes; cystathione-β-lyase (CSE), cystathionine-β-synthase (CBS), or 3-mercapto-sulphurtransferase.58 Although NO and CO are the first two identified gasotransmitters, recently, different arguments have pointed out hydrogen sulphyde (H2S) as the third gasotransmitter that can modulate vascular tone.59 Indeed, it has been recently suggested that H2S regulates HSCs contraction and that a decreased expression of CSE in HSCs may be responsible for the increased intrahepatic resistance in rodent models of liver cirrhosis.60
Increased splanchnic blood flowDevelopment of a hyperdynamic splanchnic circulatory state is a major component of portal hypertension. The increase in blood flow in splanchnic organs draining into the portal vein, and the subsequent increase in portal venous inflow, aggravates and perpetuates the portal hypertensive syndrome, especially when portal-systemic collaterals are extensive.2 The mechanisms underlying this splanchnic hyperemia are not fully understood, but it has been shown that it is associated with an overproduction of endogenous vasodilators and a decreased vascular reactivity to vasoconstrictors.2 Recent studies have demonstrated that an increased neovascularization in splanchnic organs, through a vascular endothelial growth factor (VEGF)-dependent angiogenic process, plays an important role in allowing such an increased splanchnic blood inflow, since suppression (blockade) of VEGF signalling markedly attenuates the increase in splanchnic blood flow, as well as the increased splanchnic vascularization observed in portal hypertensive animals.61,62 Interestingly, this VEGF-dependent angiogenesis also contributes significantly to the formation of portal-systemic collateral vessels in portal hypertensive animals, as described later on. Therefore, modulation of angiogenesis may represent a potential target in the treatment of portal hypertension.
Increased production of circulating/local paracrine vasodilators. Excessive response to vasodilators/Resistance to endogenous vasoconstrictorsVarious evidence suggest that there is an interrelationship between different vasoactive systems, which are coupled to cause the splanchnic vasodilation. Furthermore, it appears that none of these vasoactive factors is the only factor responsible for the splanchnic vasodilation present in portal hypertension, which is likely to be multifactorial in origin.
Circulating vasodilatorsGlucagonMany studies have demonstrated that plasma glucagon levels are elevated in patients with cirrhosis and experimental models of portal hypertension. Hyper-glucagonemia results, in part, from a decreased hepatic clearance of glucagon, but more importantly from an increased secretion of glucagon by pancreatic a cells.63 Glucagon may promote vasodilation by a dual mechanism: relaxing the vascular smooth muscle and decreasing its sensitivity to endogenous vasoconstrictors, such as norepinephrine, angiotensin II, and vaso-pressin.64,65 The role of glucagon in the splanchnic of portal hypertension provides a rationale for the use of somatostatin and its synthetic analogues to treat portal hypertension.66
EndocannabinoidsRecent data suggest a role for endocannabinoids in the hyperdynamic circulation of portal hypertension.67,68 Increased levels of endogenous cannabinoid anandamide have been found in the monocyte fraction of blood from cirrhotic humans and rats and an increased expression of the cannabinoid CB1 receptors was found in hepatic human endothelial cells.69 In addition, CB1 receptor blockade reduced portal blood flow and pressure and increased arterial pressure in cirrhotic rats.69,70 The mechanism of action is not well understood. It has been suggested that it could be due, at least in part, to an increased NO production, mediated by the activation of endothelial CB1 receptors.69 Recently, Moezi et al. have demonstrated that the role of anandamide in the pathogenesis of hyperdynamic circulation in cirrhosis are primarily mediated via stimulation of CB1 and VR1 receptor pathways.68
Several other circulating vasodilators have been implicated in splanchnic vasodilatation. Bile acids are increased in portal hypertension and have vasodilator properties. However, the data in the literature are controversial, and the role of bile acids in the hyperdynamic circulation is not well defined.71,72 Likewise, the role of the capsaicin-calcitonin gene-related peptide (CGRP) vasodilator pathway in the systemic and splanchnic vasodilatation of portal hypertension is controversial.73 Other candidates, including neuropeptides, adenosine, endotoxin, and a variety of vasodilator gastrointestinal hormones, have also been studied. However, evidence is scarce for most of them.
Local paracrine vasodilatorsMost investigators now agree on that local paracrine/autocrine vasodilators, mainly nitric oxide (NO), but also carbon monoxide (CO) and prostacyclin, play a major role in the pathogenesis of the circulatory abnormalities associated with chronic portal hypertension.
Nitric oxideExperimental studies using specific NO inhibitors have shown that NO is involved in the regulation of splanchnic and systemic hemodynamics in portal hypertensive and normal animals. The splanchnic vasoconstrictive effect caused by NO inhibitors is significantly greater in portal hypertensive than in control animals, which suggests that an excessive production of NO may be responsible, at least in part, for the vasodilatation observed in portal hypertension.74,75 In addition, an overproduction of NO has been clearly demonstrated in vitro in perfused mesenteric artery preparations from portal hypertensive rats.76 The finding in patients with cirrhosis of increased serum and urinary concentrations of nitrite and nitrate, which are products of NO oxidation, also supports a role for NO in the genesis of the circulatory disturbances of portal hypertension.77
The increased production of NO is due both to an increased expression and to an increased activity of eNOS.78,79 Factors likely to activate the constitutive eNOS include shear stress, circulating vasoactive factors (endothelin, angiotensin II, vasopressin, and norepinephrine) and overexpression of the angiogenic factor VEGF.61,62 In portal hypertensive animals NO overproduction by eNOS in the splanchnic circulation precedes the development of the hyperdynamic circulation.80
The posttranslational regulation of eNOS in portal hypertension has been further evidenced by recent studies in the partial portal vein ligated model of portal hypertension, showing that upregulation of eNOS catalytic activity, rather than eNOS overexpression, is the initial event that induces NO overproduction in the splanchnic circulation. Indeed, eNOS phosphorylation by AKT seems to be the mechanism of the initial upregulation of eNOS activity and NO-mediated hyporesponsiveness to vasoconstrictors.81 Later on, other mechanisms for an increased production of NO become important, including an enhanced signalling of the molecular chaperone heat shock protein 90 (Hsp90).82,83
Carbon monoxideRecent studies have shown an increased expression and activity of the inducible form of heme oxygenase (HO-1) in splanchnic tissues from portal hypertensive animals.84,85 In addition, the simultaneous inhibition of NO and HO has been shown to reverse completely the reduced vasoconstrictor response to potassium chloride of the mesenteric vascular bed.85
Altogether, these data suggest that the splanchnic vasodilatation present in portal hypertension is likely to be multifactorial in origin, being promoted in part by an excessive release of NO, CO and other vasoactive-mediators. In addition, experimental studies suggest that when one of the vasoactive mediators is chronically inhibited, the enhancement of other vasoactive pathways may prevent the correction of splanchnic vasodilatation.86
Hydrogen sulphyde (H2S)A growing body of recent evidence suggests that H2S is a potent vasodilator in aorta and mesenteric arteries.87 Increased activity of CSE has been demonstrated in rats with liver injury due to carbon tetrachloride.88 Recently, it has been postulated a role of H2S in the development of a hyperdynamic circulation in cirrhosis. This is based on the idea that endotoxaemia leads to increased NO synthesis and upregulation of the enzyme responsible for H2S production.
ProstaglandinsSeveral studies support a role for prostaglandins in the hyperdynamic circulation of portal hypertension.89-91 Prostacyclin is an endogenous vasodilator produced by vascular endothelial cells. It causes vascular smooth-muscle relaxation by activating adenylate cyclase and augmenting the intracellular level of cyclic adenosine monophosphate.92
COX-1 and COX-2 are involved in the biosynthesis of prostacyclin.12,17,92 It has been shown that patients with cirrhosis have increased systemic levels of prostacyclin93 and prostacyclin has also been found to be increased in the portal vein91 and aorta16 of portal-hypertensive rats. In addition, the inhibition of prostaglandin biosynthesis by indomethacin reduces the hyperdynamic circulation and portal pressure in patients with cirrhosis and portal hypertension,94 as well as in experimental models of portal hypertension,95,96 and attenuates the vascular hypore-sponsiveness to vasoconstrictors of the mesenteric vascular bed.96,97
Portal-systemic collateral circulationThe development of portal-collateral circulation is one of the main complications of portal hypertension.2 Formation of collaterals is a complex process involving the opening, dilatation, and hypertrophy of preexisting vascular channels.98 Collaterals develop in response to the increased portal pressure. A minimum HVPG threshold of 10 mmHg should be reached for the development of portal-systemic collaterals and esophageal varices.99,100
In addition to the increased portal pressure, recent studies have shown that formation of portal-systemic collateral vessels in portal hypertension is influenced by a VEGF-dependent angiogenic process, and can be markedly attenuated by interfering with the VEGF/VEGF receptor-2 signalling pathway.61,62 These studies have opened a new perspective in the understanding of the pathophysiology of portal hypertension, with potential clinical relevance, since indicate that manipulation of the VEGF may be of therapeutic value.
The collateral circulation may carry as much as 90% of the blood entering the portal system. In this circumstance, the vascular resistance of these vessels becomes a major component of the overall resistance to portal blood flow, and therefore may be very important determining portal pressure. Also, although traditionally it has been thought that the hyperdynamic splanchnic circulatory state associated with portal hypertension was the consequence of active splanchnic vasodilatation, recent data suggests that the increased neovascularization in splanchnic organs is playing an important role allowing the increase in splanchnic blood inflow.62
The elements that modulate collateral resistance are not well known. Vasoconstrictive agents (including vaso-pressin and non-selective β-blockers) may increase significantly the collateral resistance. The increase in portal collateral resistance brought about by these agents attenuates the reduction in portal pressure achieved by reducing the splanchnic blood flow.101 Another circumstance in which active changes in portal collateral resistance appear to modulate changes in portal pressure is the restitution of blood volume following haemorrhage, during which a paradoxical increase in portal pressure occurs in portal hypertensive animals.
Increased plasma volume and hyperkinetic circulationSplanchnic vasodilatation is typically associated with peripheral vasodilatation and a systemic hyperkinetic syndrome, which is characterized by reduced arterial pressure and peripheral resistance and increased plasma volume and cardiac output. The pathophysiologic mechanisms involved in peripheral vasodilatation are similar to those previously described for splanchnic vasodilatation.2 Peripheral vasodilatation plays a major role in the activation of endogenous neurohumoral systems that cause sodium retention and expansion of the plasma volume, followed by an increase in the cardiac index.102
Expansion of the plasma volume is a necessary step to maintain an increased cardiac index, which in turn aggravates portal hypertension.102 This provides the rationale for using a low-sodium diet and diuretics in the treatment of portal hypertension.
AcknowledgmentsThis study was supported by grants from the Ministerio de Educación y Ciencia (SAF:04/04783) and from the Fondo de Investigaciones Sanitarias (FIS 05/1285, 06/0623).



