




Background/Aims. This study investigated how HBV replication and host immune response are effected by reduced expression of TGF-ß1 and HBx.
Material and methods. Short interfering RNA (siRNA) knockdown technology has been used to examine the role of TGF-ß1 in hepatitis B virus replication. The siTGF-ß1 has been transfected along with 1.3mer HBV x-null to investigate the knockdown effect of TGF-ß1 on HBV replication and host immune factors.
Results. In this study, we found that diminished expression of TGF-ß1 and increased expression of HBx enhances HBV replication several folds. The differential expression of TGF-ß1 and HBx also stimulated transcriptional viral replicative intermediate (pgRNA) and secretion of core and ‘e’ antigen at translational level. Consequently, several cytokines such as IL–2, IL–8 and chemokine monocyte-chemoattractant protein (MCP–1) were increased significantly in response to stimulation of HBV replication. In contrast, TNF-α and RANTES mRNA expression increased insignificantly in response to enhanced HBV replication.
Conclusions. We concluded that reduced expression of TGF-ß1 together with HBx expression stimulate HBV replication and immune response, although the underlying mechanism of stimulation most likely differs.
Hepatitis B virus (HBV) is the prototypie member of the hepadnavirus family and is a major cause of liver disease worldwide, ranging from acute to chronic to liver cirrhosis and hepatocellular carcinoma (HCC).1–4 The distinct cell populations of the liver, namely hepatocytes, Kupffer cells, sinusoidal endothelial cells and stellate cells, are arranged in highly organized manner.5 The pathogenesis of HBV-induced liver diseases involves complicated mechanisms and implicates two important phases; viral replication and immune responses against HBV infection.6–11 During the infection of HBV, the host immune response is responsible for both hepatocellular damage and viral clearance.12,13 Although the replicative phases have been studied in detail, the immunological manifestations are not completely understood.
HBV infected hepatocytes trigger early innate defense mechanisms in order to contain viral spread, although it has no role in disease pathogenesis and viral clearance. In contrary, adaptive immune responses play vital role in pathogenesis of liver injury and clearance of virus.14–17 Virus specific CD4+ Thelper cells and CD8+ cytotoxic T lymphocytes (CTLs) contribute to tissue damage and participate in viral clearance, either by killing infected cells or by producing soluble factors, such as chemokines and cytokines.18–23 These cytokines and chemokines play regulatory role involved in immune responses against viral infection and may directly inhibit viral replication.23,24 The TGF-ß1 is a multifunctional cytokine involved in the regulation of growth and differentiation of both normal and transformed cells.24 Additionally, it plays a pivotal role in modulating the immune response through the regulation of immune cells.25 Several studies revealed that patients with chronic hepatitis and cirrhosis have significantly higher levels of plasma TGF-βΙ compared to normal subjects.26,27 Recently, it has been reported that TGF-ß1 may play an important role in regulating the progression of viral chronic hepatitis, cirrhosis, and carcinoma.26–30 Similar to TGF-βΙ, HBV X gene (HBx), a multifunctional regulatory protein are suggested to play a critical role in HBV induced hepatocellular carcinoma.31,32
Recently, Chou, et al. showed suppressed HBV replication in presence of enhanced TGF-βΙ level in the cultured cells and this decline was through preferential reduction of the synthesis of HBV pregenomic RNA (pgRNA) and core protein.29 Our previous study demonstrated that five-fold decrease in HBV replication in absence of HBx through reduction of HBV pregenomic RNA (pgRNA) level.33 These findings suggest that both TGF-βΙ and HBx regulate HBV replication via pgRNA synthesis, although at different levels of their expressions. Therefore, we evaluated in vitro the differential level of TGF-βΙ and HBx expression on the stimulation of HBV replication and host immune response elements.
Material and MethodsCell culture and transfectionHepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco-BRL) and 10 gentamicin mL–1 at 37 °C in 5% and passaged every third day. Briefly, cells were plated at 50–60% confluency. Cells were transfected using lipofectamine TM 2000 according to manufacturer’s instruction.
Plasmids and small interfering RNA (si-RNA) sequenceThe 1.3mer overlength WT HBV-X null replicon construct (containing 1.3 units of the HBV genome spanning nt 1234–1978 of HBV ayw subtype) was prepared as described previously.34 pCMV-HBx, a flag tagged HBx expression vector was generated by inserting a fragment encoding X-ORF in-frame into the p3XFLAG-CMV–14 plasmid (sigma). siTGF-β! was obtained from Dharmacon, Inc. having siTGF-b1 sense sequence CCA GAA AUA CAG CAA CAA UUU and siTGF-β! anti-sense sequence AUU GUU GCU GUA UUU CUG GUU. The scramble sequence was as follows:
- •
Sense: AAGTCGAGTCGCGTATGCAGGCCTGT CTC.
- •
Anti-sense: AACCTGCATACGCGACTCGACCCT GTCTC.
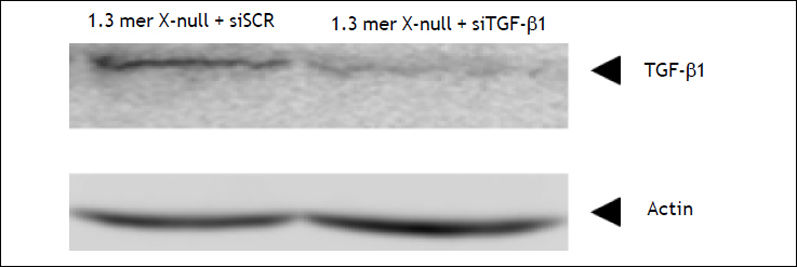
HepG2 cells were transfected with 40 nM siRNA using lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. The knockdown efficiency of siTGF-β1 was tested along with scrambled (siSCR) by western blot (Figure 1).
Transfection
To examine the effects of siTGF-β1 on HBV replication and host immune cytokines, cells were transfected in three different dishes with different combinations. For transfection, cells were grown in 100 mm dishes and transfected as follows:
- •
Dish 1: 1.3mer HBV WT X-null+ siSCR.
- •
Dish 2: 1.3mer HBV WT X-null+ HBx.
- •
Dish 3:-β1.3mer HBV WT X-null+ siTGF-β1.
Viral DNA from cytoplasmic capsids was isolated from HepG2 cells at 4 days post-transfection, as described previously.35 Southern blot analysis was performed as described previously.36 Briefly, the extracted viral DNA was separated by electrophoresis through a 1.3% agarose gel in 0.5 x Tris/acetate/EDTA buffer and transferred onto a nylon membrane. The membrane was pre-hybridized and then hybridized with a 32P-labelled full-length HBV DNA probe in hybridization solution for 16 h at 65 °C. Images were obtained using a PhosphorImager (BAS–2500; Fujifilm).
RNase protection assay (RPA)RNA extraction was performed as described above in the primer-extension assay37 and an RPA was performed essentially as described previously.38 The riboprobe was derived from the core region (nt 1903–2140) of the promoter as described previously34 Briefly, samples of total RNA or core-associated RNA (30 μg) were hybridized for 16 h at 42 °C with two probes labelled with [α-32P]UTP (3,000 Ci mmol–1; Amersham). RNase digestions were carried out with a mixture of RNase A and RNase T1 (Ambion) at 37 °C for 30 min. The digested products were separated on a 6% acrylamide/8 M urea gel, which was dried and imaged using a Phosphor-Imager.
Detection of Core and ‘e’ antigen by ELISAThe amount of HBV core (HBcAg) and ‘e’ (HBe-Ag) secreted into the culture supernatant was measured in all above experimental condition by enzyme-linked immunosorbent assay (ELISA) kit according to manufacturer’s protocol (DiaSorin, UK). Each experiment was performed in triplicate and repeated 3 times independently.
Gene expression by end point and real time PCRTotal cellular RNA from three different dishes was extracted using Trizol (Invitrogen, Life Technologies) according to the manufacturer’s protocol. Two micrograms of total RNA was used to synthesize cDNA using high capacity RNA to cDNA kit (Applied Biosystem). The end point PCR was used to check the mRNA expression of different host cytokines and chemokines. To confirm the end point PCR findings, real time PCR was performed using SYBR Green qPCR master mix method (Applied Biosystem) in a 7500 Real Time PCR System (Applied Biosystem).The experiments were repeated in triplicates. The real time primers sequence of each gene is provided in table 1.
Primer sequence and annealing temperatures used for real time PCR.
| Primer name | Primer sequence | Annealing (°C) |
|---|---|---|
| MCP1-FMCP1-R | ACTCTCGCCTCCAGCATGAATTGATTGCATCTGGCTGAGC | 60 |
| IL–8 FIL–8 R | CTGCGCCAACACAGAAATTATTGTATTCACTGGCATCTTCACTGATTCTT | 63 |
| IL–2-FIL–2-R | TGCACATAATCTCATCTTTCTAACACTCTTTTGAAAGCGCAATAGATGGACAT | 61 |
| RANTES-FRANTES-R | TTTGCCTACATTGCCCGCTTTCGGGTGACAAAGACGACT | 58 |
| TNFα-F TNFα-R | CCCAGGGACCTCTCTCTAATCA GCTTGAGGGTTTGCTACAACATG | 59 |
A two tailed Student’s t-test, assuming unequal variances, was used for all statistical analyses; a P value of < 0.05 was considered statistically significant.
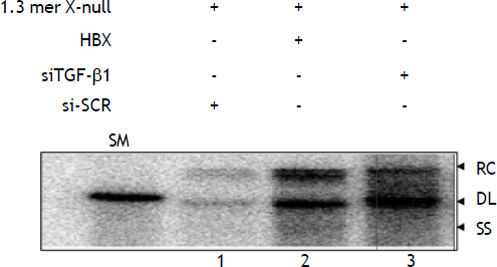
ResultsTGF-βΙ knockdown enhances HBV replicationTo determine the effect of TGF-β1 knockdown on HBV replication, HepG2 cells were co-transfected with 1.3 mer HBV X-null replicon along with siT-GF-β1 and siSCR. Southern blot analysis indicated that TGF-β1 knockdown significantly enhances HBV replication (Lane 3) compared to scrambled (Figure 2, lane 1 vs. lane 3). The fold increase in HBV replication by TGF-β1 knockdown was almost similar to our previous finding, when HBx was complemented with 1.3 mer HBV X-null replicon in HepG2 cell (Figure 2, lane 1 vs. lane 2). Our results indicated that the regulatory mechanism of TGF-ß1 and HBx might have antagonistic mode of action. Therefore, to prove this antagonistic mode of action at transcriptional level, pgRNA level was measured by RNase protection assay.
 1.3 mer HBV x-null + siSCR. ii) 1.3 mer HBV x-null + HBx. iii) 1.3 mer HBV x-null + siTGF-β1. Southern blot was performed as discussed in materials and methods. The replicative DNA intermediates are: RC: relaxed circular DNA. DL: duplex linear DNA. SS: single-stranded DNA. A restriction fragment representing one HBV genomic unit (3.2 kb) served as a size marker (SM). The value for lane 1 (WTX-null + siSCR) was set as 100.")
Activation of HBV replication by siTGF-βΙ and HBx in HepG2 cell. HepG2 cells were transfected in three 100 mm plate with different combinations: i) 1.3 mer HBV x-null + siSCR. ii) 1.3 mer HBV x-null + HBx. iii) 1.3 mer HBV x-null + siTGF-β1. Southern blot was performed as discussed in materials and methods. The replicative DNA intermediates are: RC: relaxed circular DNA. DL: duplex linear DNA. SS: single-stranded DNA. A restriction fragment representing one HBV genomic unit (3.2 kb) served as a size marker (SM). The value for lane 1 (WTX-null + siSCR) was set as 100.
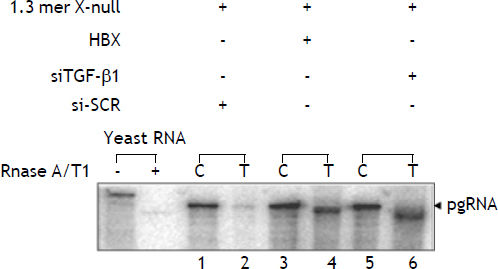
Previously, we reported that wild type HBx complemented with 1.3 mer HBV X-null facilitates viral pregenomic RNA (pgRNA) transcription by approximately five fold compared to full length HBV x-null construct. To clarify, whether knockdown of TGF-ß1 resulted in pgRNA stimulation at transcriptional level, RNase protection assay was performed. Total and capsid-associated viral transcripts were isolated from the cytoplasm. RPA of total cytoplasmic RNA revealed that the amount of pregenomic RNA from cells transfected with 1.3 mer HBV X-null+ siTGF-ß1 was enhanced by approximately fivefold relative to that of the 1.3mer HBV X-null +siSCR (Figure 3, lane 1 and 2 vs. lane 5 and 6). Thus it was consistent with the interpretation that TGF-ß1 knockdown contributes to viral genome replication entirely at the transcriptional level and hence antagonistic to HBx action.
 1.3 mer HBV x-null + siSCR. ii) 1.3 mer HBV x-null + HBx. iii) 1.3 mer HBV x-null + siTGF-ß1. RNA extracted from the capsid (C) and total cytoplasmic (T) fractions was analyzed by RNase Protection Assay (RPA). The RPA probe was derived from the C gene region for specific detection of pgRNA. Quantification was estimated by comparing the amount of pgRNA in the capsid fraction to that found in the total cytoplasmic fraction.")
Upregulation of pregenomic RNA by knockdown of TGF-ß1 and overexpression of HBx. HepG2 cells were transfected in three 100 mm plate with different combinations: i) 1.3 mer HBV x-null + siSCR. ii) 1.3 mer HBV x-null + HBx. iii) 1.3 mer HBV x-null + siTGF-ß1. RNA extracted from the capsid (C) and total cytoplasmic (T) fractions was analyzed by RNase Protection Assay (RPA). The RPA probe was derived from the C gene region for specific detection of pgRNA. Quantification was estimated by comparing the amount of pgRNA in the capsid fraction to that found in the total cytoplasmic fraction.
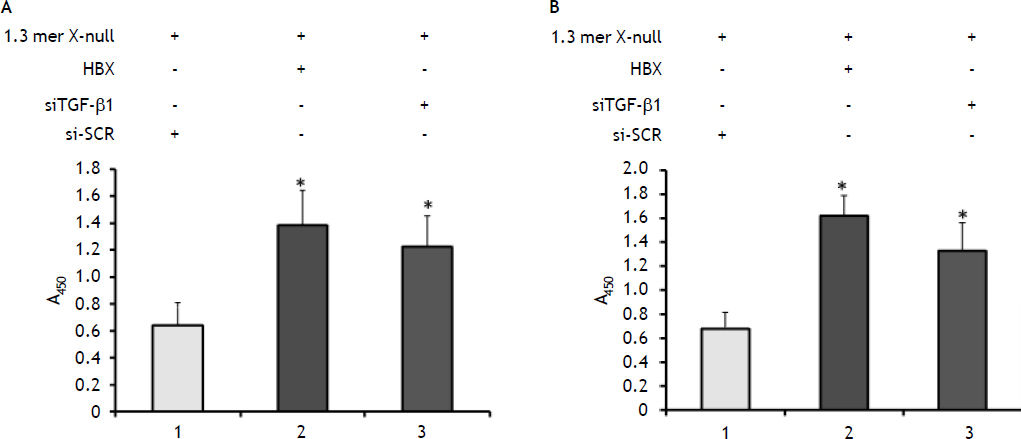
We then assessed the impact of TGF-ß1 knockdown on the secretion of core and ‘e’ antigen into culture medium of cells by commercially available ELISA kit. This was done in order to find the level of core and ‘e’ antigen secretion from transfected 1.3 mer HBV X-null in presence of siTGF-β1 and HBx. Consistent with the above transcriptional level data, both core and ‘e’ antigen secreted in culture medium were increased to 2–3 fold in presence of siTGF-β1 and HBx (Figures 4A and 4B).
 and HBeAg (B) secretion by knockdown of TGF-ß1 and overexpression of HBx. The amount of HBcAg and HBeAg secreted into the culture supernatant was measured using a commercial available ELISA kit according to the manufacturer’s instructions (Daisorin). The absorbance was measured at 450 nm as shown in figures 3A and 3B. All data were presented as mean ± SEM of triplicate values from three independent experiments. Significant difference from control (1.3mer HBV x-null + siSCR) is represented by an asterisk (*P < 0.05).")
The impact on HBcAg (A) and HBeAg (B) secretion by knockdown of TGF-ß1 and overexpression of HBx. The amount of HBcAg and HBeAg secreted into the culture supernatant was measured using a commercial available ELISA kit according to the manufacturer’s instructions (Daisorin). The absorbance was measured at 450 nm as shown in figures 3A and 3B. All data were presented as mean ± SEM of triplicate values from three independent experiments. Significant difference from control (1.3mer HBV x-null + siSCR) is represented by an asterisk (*P < 0.05).
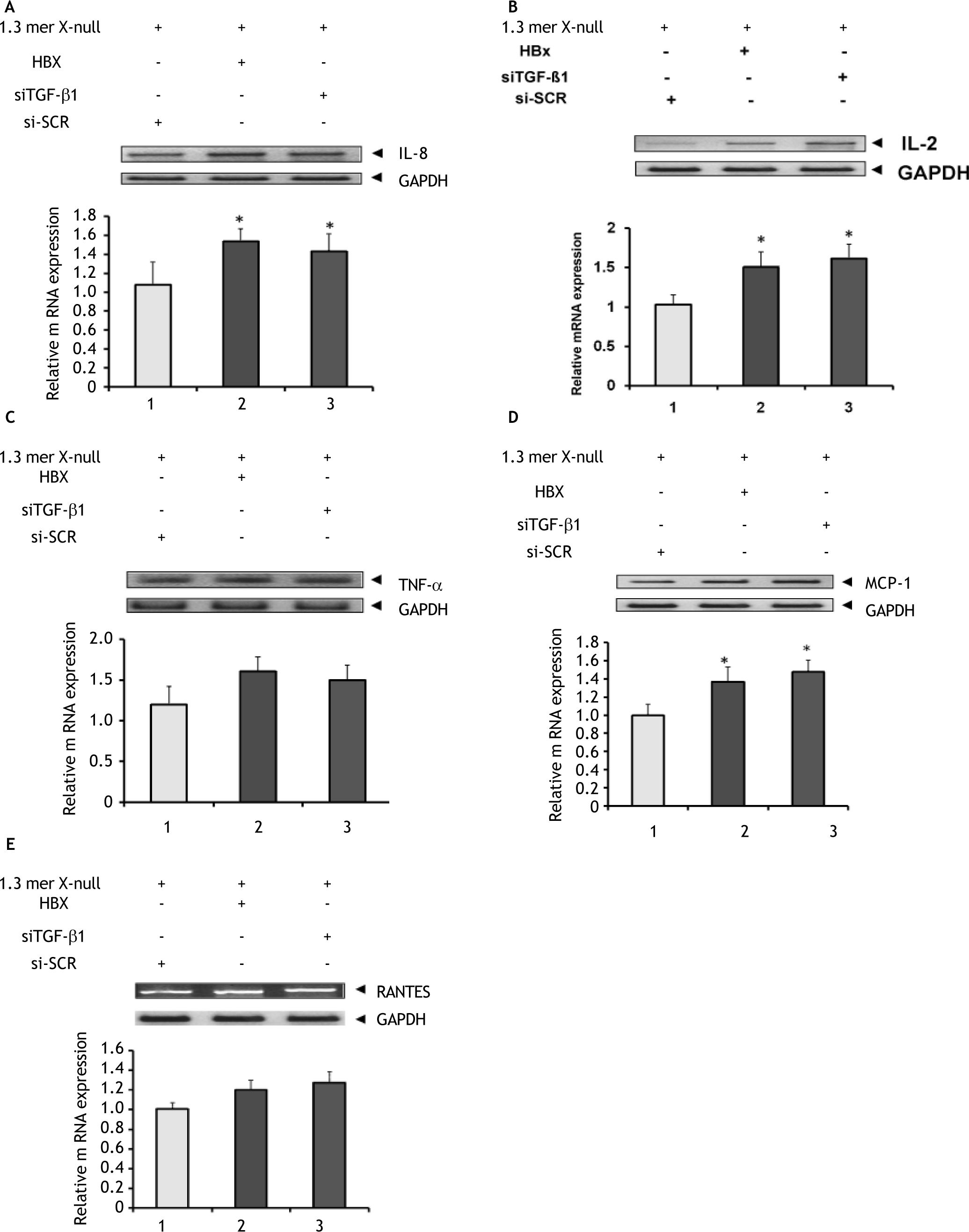
TGF-β1is a secreted protein that performs many cellular functions and are involved in stimulatory as well as inhibitory effects in different cell types. It also play an important role in regulating the immune mechanism of the host cells. Therefore we tested; if knockdown of TGF-β1 expression in HBV replicating cells affect the level of immune cytokines and chemokines mRNA (such as IL-8, IL-2, TNF-α, RANTES and MCP-1) that regulates viral clearance. Reverse transcription-PCR (end point PCR) was performed using specific primers for each gene to identify the mRNA expression level and were reconfirmed by real time quantitative PCR. As shown in figures 5A and 5B, siTGF-β1 and HBx induced expression of both interleukin–8 (IL–8) and interleukin-2 (IL-2) to a significant level. Similarly, siTGF-β1 and HBx action upregulated the mRNA expression of TNF-α, but to a insignificant level compared to 1.3mer X null+ siSCR transfected (Figure 5C). The expression of the monocyte-chemoattractant protein (MCP-1), a chemokine increases to a significant level in presence of siTGF-β1 and HBx compared to1.3mer X null + siSCR (Figure 5D), while another important chemokine (RANTES) level increased but insignificantly (Figure 5E).
 and experimental groups were analyzed for the expression of different cytokines and chemokines by end point RT-PCR and reconfirmed by real time PCR as shown in figure 4A–4E. All data were presented as mean ± SEM of triplicate values from three independent experiments. Significant difference from control (1.3mer HBV x-null + siSCR) is represented by an asterisk (*P < 0.05).")
Effect of TGF-ß1 knockdown on gene related to cytokines and chemokines. HepG2 cells were transfected in three 100 mm plate with different combinations as discussed in figure legend 1. Total cellular RNA was isolated with Trizol. RNA from control (1.3mer HBV x-null + siSCR) and experimental groups were analyzed for the expression of different cytokines and chemokines by end point RT-PCR and reconfirmed by real time PCR as shown in figure 4A–4E. All data were presented as mean ± SEM of triplicate values from three independent experiments. Significant difference from control (1.3mer HBV x-null + siSCR) is represented by an asterisk (*P < 0.05).
Recent studies have revealed that patients with chronic hepatitis and cirrhosis-HCC have significantly higher levels of plasma TGF-β1 than do normal subjects.7,39–41 Chou, et al. reported that the exogenous addition of TGF-β1 effectively suppress HBV replication in cultured liver cells.29 This suppression was due reduction in pgRNA at the transcriptional level. It is known that pgRNA encodes the core/polymerase proteins and is substrate for viral encapsidation. Therefore, expression of pgRNA directly affects the level of viral replication. This lead us to analyze how knockdown of cytokine TGF-β1 affect HBV replicating HepG2 cells at viral replication, transcription and translational level as well how it stimulates host cytokines and chemokines involved in viral clearance. Our results showed that the low expression of TGF-β1 (siTGF-β1) enhanced HBV replication compared to higher expression of this cytokine (siSCR). Our result was consistent with the earlier findings that enhanced TGF-β1 level might directly inhibit HBV replication.29,30 Additionally, the complementation of HBx and 1.3 mer HBV X-null construct enhances HBV replication by several folds.33 Hence, the concentration of HBx and TGF-β1 in hepatocytes plays a vital role in the stimulation of HBV replication. We speculated that like HBx, TGF-β1 might also be involved in regulating number of cellular and viral genes in HBV infected cells.
Furthermore, our result demonstrated that decreased TGF-β1 level enhances HBV replication through transcriptional upregulation of pregenomic RNA. These complement earlier findings which indicated that the level of TGF-β1 regulates pgRNA synthesis primarily through transcriptional regulation.29,30 Apart from regulation of pgRNA by TGF-β1 we also have reaffirmed the role of HBx in HBV replication through transcriptional upregulation of pregenomic RNA. This corroborate earlier findings that enhancement of the pgRNA occurred at the transcriptional level.33,34 Hence, this study proves the notion that TGF-β1 and HBx stimulate viral genome replication at the transcription level. In order to find how TGF-β1 effect the synthesis and secretion of HBcAg and HBeAg, enzyme linked immunosorbent assay (ELISA) was performed. In contrast to high level of TGF-β1 low level of TGF-β1 enhances core and ‘e’ antigen synthesis. Similarly, HBx also augments core and ‘e’ antigen synthesis; strengthen the notion that TGF-β1 and HBx play a vital role in replication, transcription and translation of HBV.29,33,34 These confirm earlier findings that both TGF-b1 and HBx transactivate HBV core promoter.29,33,34 Our result showed similar effect of TGF-β1 on both ‘core’ and ‘e’ antigen and hence contradict earlier findings that TGF-β1 inhibit the synthesis of pre-C mRNA to a lesser extent.29,33
We have also analyzed the expression of some important cytokines and chemokines responsible for the immunity and inflammation in presence of viral particles. Recently, several studies reported the role of cytokines and chemokines in effective suppression of HBV replication.8–10 The knockdown of TGF-β1 significantly enhanced the expression of cytokines IL-2 and IL-8 and chemokines MCP-1, when compared with knockdown (minus) Huh7 cells. The expression level of TNF-a and RANTES too increased in TGF-ß1 knockdown cells but not to a significant level. The level of cytokines and chemokines imitate TGF-ß1 knockdown cells, when HBx complement with 1.3 mer HBV X-null. The augmented cytokines level may be due to increase HBV replication in knockdown cells, and are likely to be involved in the regulation of immune responses against viral infection and may directly inhibit viral replication.42 Therefore, in absence or low level of TGF-ß1 may trigger cascades of antiviral responses involving cytokine and chemokines. Although, HBx increases HBV replication by several folds, we speculate that it has adjunct role in chronicity. The host defence machinery (including chemokines and cytokines) play a vital role in determining the viral clearance and persistence.
ConclusionThe differential expression of TGF-ß1 and HBx stimulate HBV replication and immune response, although the underlying mechanism of stimulation most likely differs. We speculate that the activity of TGF-ß1 and HBx vary among different hosts and hence the disease outcome.
AcknowledgementThe authors extend their appreciation to the Centre of Excellence in Biotechnology Research (CEBR) at King Saud University, Riyadh, Saudi Arabia, for funding the work through small projects (opportunity-fund). The authors also extend their deepest gratitude to Dr. Wang Shick Ryu, Department of Biochemistry, Yonsei University, Seoul 120–749, Republic of Korea for plasmid expression of 1.3mer HBV WT X-null and HBx as a kind gift.
Declaration Of InterestThe authors report no declaration of interest. Both authors contributed equally.



