



Hepatitis B viral (HBV) infection is the commonest cause of hepatocellular carcinoma. HBV DNA is the most important predictor of hepatocarcinogenesis in HB surface antigen positive patients. We reviewed the mechanism of hepatocarcinogenesis on molecular level with a special emphasis on the role of X-protein. Hepatitis B X-protein communicates with host targets and disturbs cellular functions including cell cycle regulation, apoptosis, signalling, transcriptional regulation, encoding of cytoskeleton, cell adhesion molecules, oncogenes and tumour suppressor genes.
More than 80% of hepatocellular carcinoma (HCC) is found in hepatitis B virus (HBV) infected patients.1 HCC is the fifth most common cancer in the world killing 300,000-500,000 people every year.1,4 Of approximately 2 billion infected people worldwide more than 400 million are chronic carrier of HBV.2 About 75% of chronic carriers live in Asia and western pacific.1,3 In low endemic areas HBV infection is common in injection user, homosexual men, health care workers and in patients who require regular blood transfusion or haemodialysis. In high endemic areas it is most commonly transmitted perinatally.1
The earlier person is infected the more chances that they will become chronic carrier e.g 90% in perinatal infection while 10% in adults.2 Exposure to hepatotoxins, such as aflatoxin and alcohol in person already infected with HBV hasten the development of cirrhosis and HCC.4 Coinfection with hepatitis C also doubles the risk of developing HCC.4 Prognosis is poor with median survival of less than 6 months as most of the patients have advanced and unresectable disease on presentation.
Relative risk of HCC in hepatitis e-antigen (HbeAg) positive patient is much higher than among inactive hepatitis B surface antigen (HbsAg) carriers. It can occur in convalescent HBV infection. Even patients with hepatitis B core (HBc) antibody or hepatitis B surface antibodies are at increase risk of developing HCC but much lower than those with active infection.5,6 HBV DNA is the most important predictor of HCC development in HbsAg positive patients.
Chang et al. from Taiwan showed a decline in HCC in children after universal hepatitis B vaccination.7 Another study in Korea suggested immunization even in adulthood could reduce the risk.8 Perz et al. concluded that HBV and HCV infection account for majority of HCC throughout most of the world, highlighting the need for programs to prevent new infections and provide medical management for those already infected.
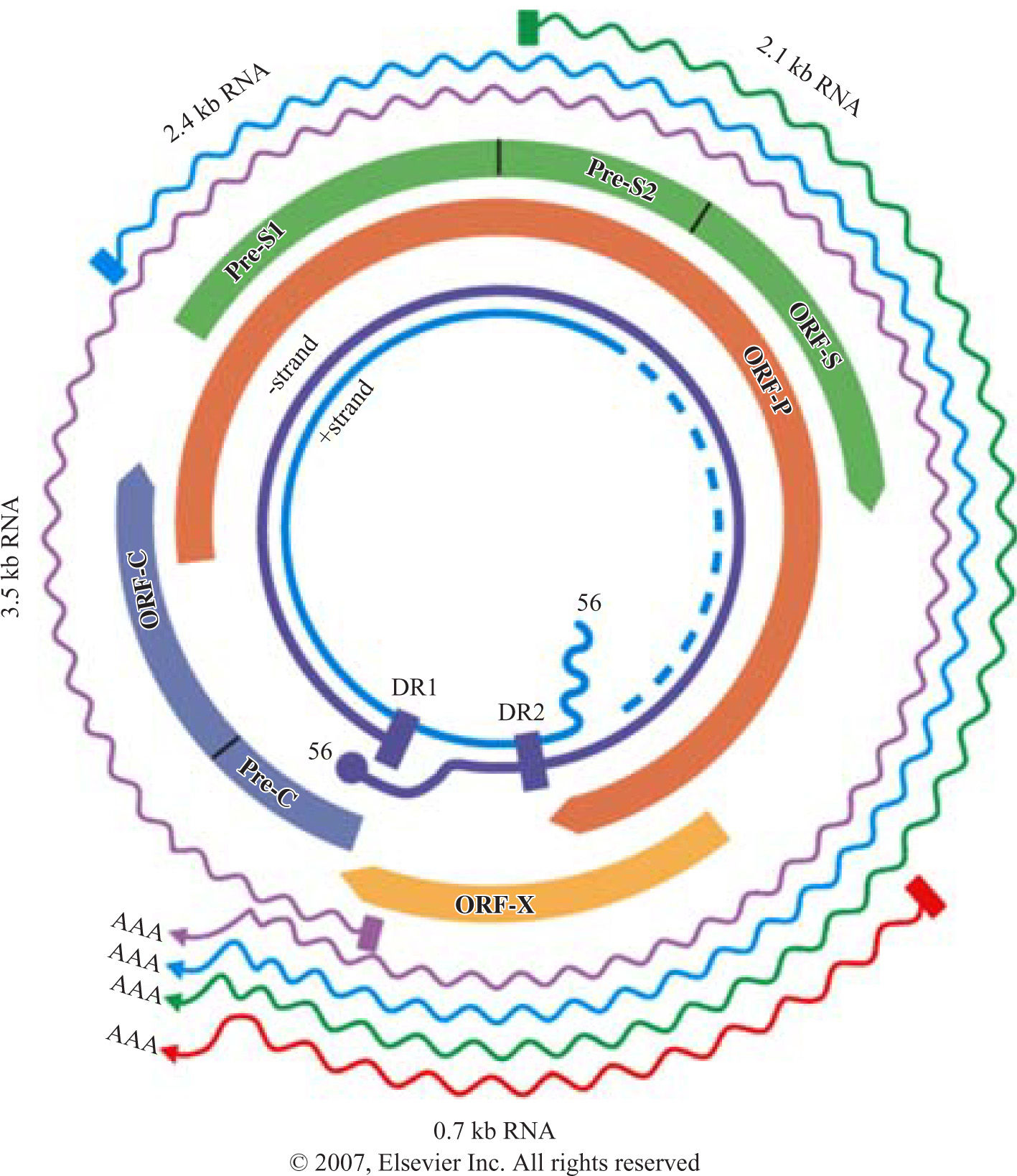
Structure of HBV genomeHBV belongs to family of hepadnaviruses. HBV genome is circular, partially double stranded DNA of 3.2 kb in length. The genome contains 4 open reading frames encoding the envelope (pre-S/S), core (pre core/core), polymerase, and X proteins (X). The pre-S/S open reading frame encodes the large (L), middle (M) and small (S) surface glycoprotein. The precore/core frame is translated into a precore polypeptide, which is modified into soluble proteins, HbeAg and the nucleocapsid protein, hepatitis B core antigen. The polymerase protein functions as a reverse transcriptase as well as a DNA polymerase. The X protein is a transactivator and may play a role in liver carcinogenesis (Figure 1).
Mechanism of carcinogenesis")
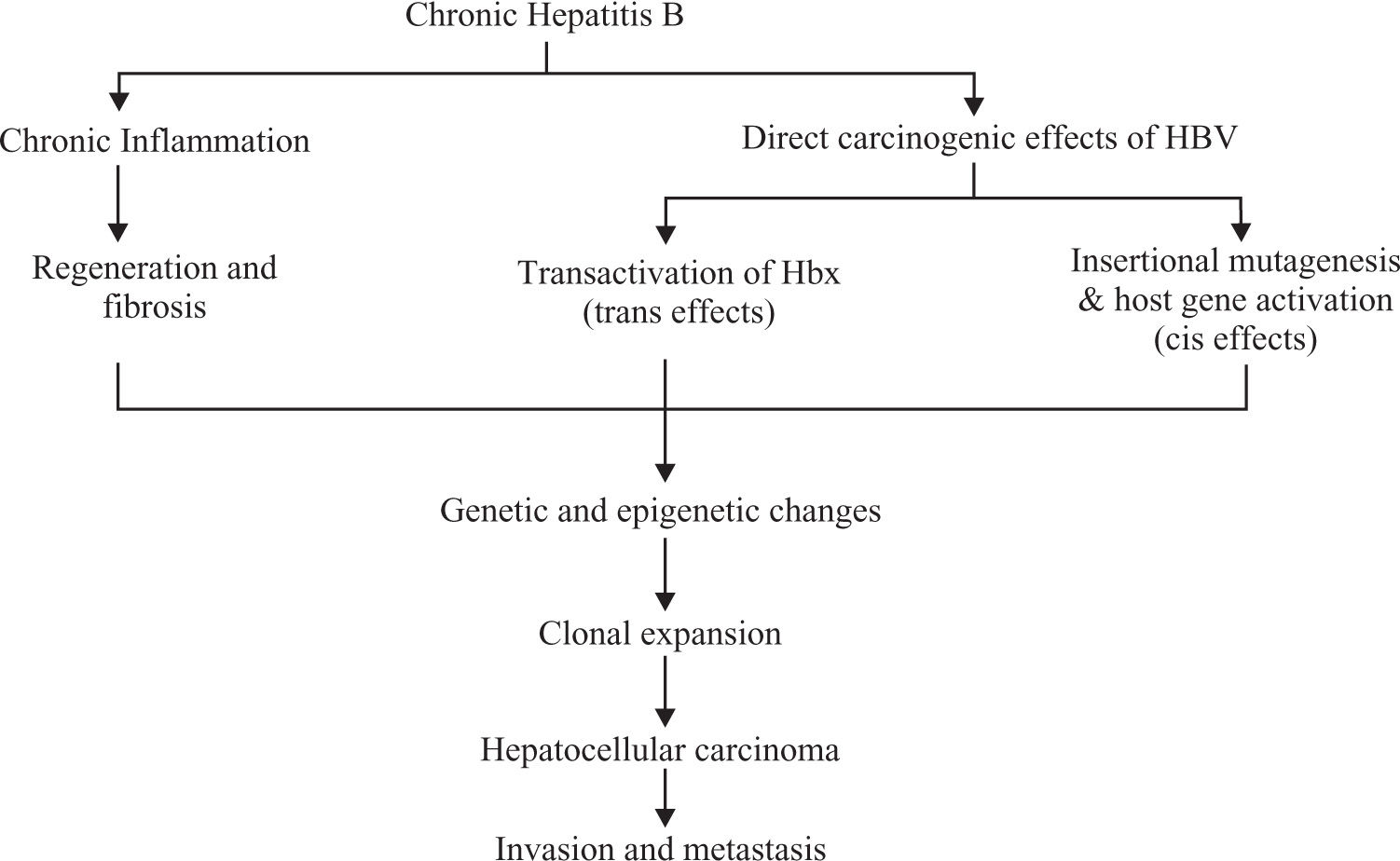
The exact mechanism is still not clear. It is thought there are 2 pathways through which HBV causes cancer.
- •
Chronic necro-inflammation of liver cells, cellular injury, mitoses and liver cell regeneration. This process leads to accumulation of mutations (Figure 2).9
- •
Direct oncogenic effect of HBV through chromosomal integration (cis activation) or transactivation of cellular genes.10 Persistent replication is associated with a high frequency of integration of HBV sequences into the human host genome5,10(Figure 2)
.")
When HBV integrations occur they produce many genetic changes in the host genome, including deletions, translocations, the production of fusion transcripts, and generalized genomic instability.3 Chromosomal losses are present in 25%-45% of patients in 1p, 4q, 5q, 6q, 8p, 9p, 13q, 16p, 16q and 17p while gains occur in 30%-55% of patients in 1p, 6p, 8q, and 17q.3,11 Many of these chromosomal segments contain tumour suppressor genes like p53, Rb, cyclinDl, p16.3
Seventy percent of the HBV associated HCC produce HBV X protein (HBx), transcriptional trans-activator. Integrated HBV sequences have carboxy-terminal deleted X gene resulting in translation of HBx protein.12 These mutants lose their capacity for oncogene induced apoptosis and cell cycle arrest, which leads to unrestricted cell proliferation that increases the transforming capacity of the protein.13
Another HBV gene that has transactivational property is a truncated form of the pre-S2/S gene.5 Pre S2/S activates mitogen activated protein kinases (MAPK/ERK) that further activates transcription factors like AP-1 and nuclear factor-***entity*** (NF-***entity***) and this causes increased proliferation of liver cells.14 Overproduction of pre- S2/S protein, particularly L, results in intracellular accumulation, which in turn cause cell to stress that results in HCC.15
HBV DNA integration into the cellular genes is frequently detected in HBV positive HCC. This viral integration leads to target gene transcription. Analysis of HBV DNA site in liver cancer enabled researchers to identify several cancer related genes and pathways. Most of the viral integration genes are in the vicinity of cellular genes or inside the coding sequence, this cause activation of expression of proto oncogene or inactivation of tumour suppression genes especially p53. Studies have shown that hTERT (human telomerase reverse transcriptase) is a recurrent site of viral integration. Studies also supported the view of recurrent targeting of genes involve in cell signaling and growth control, h- TERT, PDGF (Platelet derived growth factor) receptors, calcium signalling related genes, MLL (mixed lineage leukaemia) and 60s ribosomal protein genes.16 The h-TERT gene expression is increased in HCC.
In summary, viral integration occurring in acute phase of hepatitis induces genetic changes in target cellular genes. The oncogenic activity of cellular and viral genes- modified by the viral integration- may provide the cells having the HBV DNA integration with a selective growth advantage during the chronic phase of hepatitis. Increasing accumulation of genetic changes during liver cell proliferation leads to HCC. Sixty-one genes have been identified at HBV DNA site, which are involved in control of cell proliferation and survival and likely to play a role in hepatocarcinogenesis16.
HBx and hepatocellular carcinomaHBx communicates with variety of host targets and mediates many opposing cellular functions, including cell cycle regulation, function in apoptosis, signalling, transcriptional regulation, encoding of cytoskeleton, cell adhesion molecules, oncogenes and tumour suppressor genes.10,18
HBx up-regulates the expression of proto-oncogenes such as c-myc and c-Jun, transcription factors like NF- ***entity***B (nuclear factor kappa B), AP-1, AP-2, RPB5 subunit of RNA polymerase 2, TATA binding protein, c-Amp response element binding protein (CREB) and other viral genes like HBV enhancers in the nucleus. Target proteins that directly interact with HBx are Smad4 and nuclear factor activated T cells (NF-AT) in TNF***entity***signalling and COX-2,19 retinoid X receptors in phosphoe- nolpyruvate carboxykinase expression. Host genes indirectly affected by HBx are the up regulation of inter- leukin6 (IL6), IL8, Fas ligand and induction of nitric oxide synthetase. HBx activates MAPK/ERK, stress activated protein kinase/Jun N-terminal kinase (SAPK/ JNK) and protein kinase C (PKC) signalling pathway to regulate NF-***entity***B and AP-1 dependent transcription. Induction of NF-***entity***B and AP-1 activity lead to acceleration of cell cycle progression, increased proliferation and repression of apoptosis.17
HBx effects the signalling pathways like MAPK/ERK, protein kinase B (PKB), PKC, SAPK/JNK, Phosphatidylinositol 3-Kinase (PI-3-K) and janus kinase (JAK)/STAT kinase pathways.17
HBx can bind to C-terminus of p53 forming a proteinprotein complex and inactivating many functions of p53, including apoptosis.20 A p53 mutation is present in 30-60% of HCC patients.3 HBx sequester p53 in the cytoplasm and prevents it from entering the nucleus.21,22 HBV protects itself from apoptotic death through an HBx-P13K-AKT-Bad pathway and by inactivating caspase 3 activity that is partially p53 independent in hepatocytes.23 The proapoptotic action of HBx overcomes the inhibitory effect of Bcl-2 against Fas cytotoxicity.24 HBx also up regulates survivin expression, an inhibitor of apoptosis (IAP) family.25 HBx also promotes the apoptosis of liver cells by upregulating the expressions of Fas/Fasl, Bax/Bcl-2, Bcl-xL, and c-myc in a dose dependant manner.10,26 Thus HBx has function in both pro-apoptosis and anti apoptosis pathways dependent on different cell settings. Increased anti-apoptosis and decreased pro-apoptosis in HCC is an important mechanism.
El Far et al. showed that detection of serum p53 levels increased the frequency of HCC prediction; significant positive correlation was also found between p53 and tumour size.27 Another study showed no definite correlation between p53 positivity and tumour size, histological grade or vascular invasion.28
HBx stimulates cell cycle progression by accelerating transit through G1/S and G2/M check points. HBx increases the rate and level of activation of cyclin dependant kinase (CDK2) and their respective association with cyclins E and A or B, which are important in control of cell cycle regulation. It also over expresses cyclin D1.29
HBx induces angiogenesis through up regulation of vascular endothelial growth factor (VEGF), which is an important factor in metastases. It also inhibits the repair of damaged liver cells DNA by interacting with p53 or through binding to the damaged DNA binding protein, which is implicated in DNA repair and cell cycle regulation. This leads to accumulation of DNA mutations and cancer.
ConclusionHBV is a recognized cause of carcinogenesis. It acts as a co-factor with others like aflotoxins in hepatocarcinogenesis. Viral trans-activating genes inactivate tumour suppressor genes or activate the expression of proto-oncogenes. This causes over proliferation of virally infected cells. HBV also blocks apoptosis.
HBx is a transactivator protein in HBV genome, which through many different mechanisms causes HCC. Vaccination significantly reduces the incidence of HCC. Recent studies have given more information about the mechanism of oncogenes and tumour suppressor genes in hepatocarcinogenesis, together with molecular pathway of the control through which their effects on proliferation are mediated.



