



Considerable expectations to prevent hepatocellular carcinoma (HCC) appearance are connected with the use of Interferon α (IFN α) in antiviral treatment of hepatitis B or C. Several studies have reported that the incidence of HCC may be reduced after IFN therapy in patients with chronic B or C hepatitis although its real preventive effect is still debatable. The purpose of the studies from our laboratory was to evaluate the action of IFN a2b on preneoplastic foci in a two-phase model of preneoplasia development in rat. We demonstrated that IFN-α2b administration significantly decreased both number and volume percentage of altered hepatic foci (AHF). This reduction could be explained by an induced programmed cell death in the foci. This apoptotic effect of IFN-α2b on preneoplastic liver foci was mediated by the production of endogenous TGF/31 from hepatocytes acting by a paracrine/autocrine way. Further studies confirmed that these results were a consequence of the perturbation of the redox status induced by the IFN-α2b. In conclusion, IFN-α2b could enhance the proapoptotic effects of TGF/β1 in early stages of hepatocarcinogenesis, which could be highly beneficial in cancer therapy.
Abbrevations
IFN, Interferon
Jak, Janus-activated kinase
Stat, signal transducers and activators of transcription
NK, natural killer cells
HCC, hepatocellular carcinoma
AHF, altered hepatic foci
DEN, diethylnitrosamine
2-AAF, 2-acetylaminofluorene
TGF-ß, transforming growth factor-ß
M?s, macrophages
ROS, reactive oxygen species
??, transmembrane potential
GSH, glutathione
CAT, catalase
SOD, superoxide dismutase
ASC, ascorbic acid
InterferonsInterferon was discovered by Isaacs and Lindenmann in 1957 as a result of study of the phenomenon of virus interference. They used chick membranes suspended in a simple medium and infected with influenza viruses, and found that those cells incubated with inactivated virus released into the medium a substance which rendered other cells resistant to infection with a live virus. This mediator of interference they showed to be a protein and not a virus particle and they named it interferon (IFN).1,2
Subsequent work has shown that there are families of interferons, differing between species and also within an animal according to the cell that produces it: IFN α from virus-challenged blood leukocytes, IFN β from fibroblasts and IFN γ immune interferon from transformed lymphocytes.3
More recently, IFNs were divided into two major subgroups by virtue of their ability to bind to common receptor types.4,5 Type I IFNs all bind to a type I IFN receptor, and include IFN α, IFN β, IFN ω and IFN τ. IFN γ is the sole type II IFN, and binds to a distinct type II receptor.6
For the tipe I IFNs, there are two receptor subunits known, IFNAR-1 and IFNAR-2, which bind the Janus-activated kinase (Jak) molecules Tyk2 and Jakl, respectively. For INF y, there are two receptor subunits known: IF-NGR-1 and IFNGR-2, which associate with Jakl and Jak2, respectively. Upon binding of IFN to its receptor, the receptor undergoes oligomerization, with transphosphorylation of Jaks followed by phosphorylation of the cytoplasmic tails of the receptor molecules by the activated Jaks. This provides a docking site for the signal transducers and activators of transcription (Stats) which are then phosphorylated by the Jaks. The phosphorylated Stat dimmers are released from the receptor molecules and translocate to the nucleus, where they activate transcription of IFN-stimulated genes (ISGs). In the case of type I IFNs, ISGs can be identified by the presence of an IFN-stimulated response element (ISRE) in their promoter regions. Enhancers of INF y-inducible genes contain a unique element called the INF y activation site (GAS).7
Almost all cell types produce type I IFNs. The prototypical production sites for INF α and IFN β are leukocytes and fibroblasts, respectively. Their induction usually follows exposure to viruses, double stranded RNA, polypeptides, and cytokines.8
The type II IFN γ is produced in T cells and natural killer (NK) cells following a number of immunological stimuli, inducing T cell-specific antigens, staphylococcal enterotoxin A, and the combination of phytohemagglutinin and phorbol ester.7,8 Unlike INF α and IFN β, it is not directly induced in cells following viral infection.
Although type I IFNs have long been considered only as potent antiviral proteins, today there are unanimous recognition that they participate vigorously in the complex cytokine network that regulates differentiation, function and homeostasis of a variety of cell lineages. Type I IFNs are constitutively expressed at low levels in hematopoietic tissues where they may contribute to lymphocyte homeostasis, they are secreted in response to viral infection and upon stimulation of several Toll-like receptors (TLRs).9 In the course of an immune response, these cytokines can affect virtually all immune cells: they activate dendritic cells, enhance NK cell citotoxicity, promote T lymphocyte differentiation and affect survival of memory T cells. It is thought that collectively these immunoregulatory effects of IFN contribute to the elimination of a large variety of pathogens as well of incipient tumors.10
Interferon α and liverHuman lymphoblastoid IFN α has been shown to have a powerful antiproliferative effect on human hepatoma cell line PLC/PRF/5 in a dose-dependent manner, both in vitro and in vivo, after implantation in nude mice.11 Moreover, IFN α inhibits liver regeneration by decreasing DNA and total protein synthesis.12,13
IFN ± has antitumor activity against a variety of tumors, mainly among hematologic malignancies.14 Accordingly, considerable expectations to prevent hepatocellular carcinoma (HCC) appearance are connected with the use of IFN ± in antiviral treatment of hepatitis B or C. Several studies have reported that the incidence of HCC may be reduced after IFN therapy in patients with chronic B or C hepatitis15,16 although its real preventive effect is still debatable.17 Conversely, the benefit derived from IFN ± treatment of established HCC remains controversial.18,19 On this regards, it has been stated that it is highly likely that IFN α applied in the early stages of tumor evolution could have a very important clinical effect, whereas its activity in advanced stages in which multiple genetic aberrations are present, would be minimal.20
Liver tumorsHCC is a malignant tumor that arises from the major cell type in the liver: the hepatocyte. HCC is the most primary hepatic tumor, represents approximately 6 per cent of all malignancies and the fifth most common tumor worldwide.21
Nearly all types of primary liver tumors known to occur in humans can be reproduced by chemicals in laboratory animals, specially in rats.22 In experimental carcinogenesis, preneoplastic foci of altered hepatocytes (AHF) emerge weeks or months before the appearance of hepatocellular adenomas and HCCs23,24 and this has also been discovered in human with hepatocellular neoplasms and/ or cirrhosis.25 This fact has led to the development of a number of in vivo systems for the study of early neoplasia in rat liver.26,27 The initiation-promotion or two-stage model of cancer development mimics the early events of the latent period of human carcinogenesis. Several two-stages models have been developed, including the protocols of Solt-Farber,28 Ito et al.29 and Rao et al.30 involving necrogenic doses of carcinogens or other models such as the protocols of Peraino et al.31 and Pitot et al.32 that use low, non toxic doses of carcinogens.
The initiation stage of cancer development can be produced in rat liver by the administration of diethylnitrosamine (DEN),28-30 a complete carcinogen that produces DNA ethylation and mutagenesis.32 Necrogenic doses of DEN cause massive hepatic necrosis followed by regeneration24 and would be expected to cause not only increased gene expression related to regeneration, but also increased expression related to oncogene mutation. Administration of promoting agents causes selective enhancement of the proliferation of initiated cell populations over non-initiated cells in the target tissue.33
Therefore, the purpose of early studies from our laboratory34 was to evaluate the action of IFN α on preneoplastic foci in a two-phase model of preneoplasia development in rat.
The dose of IFN used (6.5 x 105 U/kg b.w., administered i.p. three times a week) was comparable to that used for therapeutic purposes. The INF α used was IFN α-2b, produced by recombinant technology, and generously gifted by Bio Sidus, S.A. The currently available preparations of IFN α are: IFN α-2a, IFN α-2b, IFN alfacon-1, IFN α-n3.7
We studied the effect of IFN-α2b during initiation with a complete carcinogen (DEN) and during the administration of a promoting agent (2-acetylaminofluorene, 2-AAF) on number of AHF per liver and volume fraction of the liver occupied by AHF.
To our knowledge, this was the first study that had evaluated the action of IFN-α on preneoplastic foci in vivo, by using a two-phase model of cancer development in rats. We demonstrated that IFN-α2b administration significantly decreased both number and volume percentage of AHF. The reduction of both number and volume percentage of AHF in IFN-α2b-treated animals may be explained by a greater programmed cell death in the foci.
Programmed cell deathApoptosis, or programmed cell death, is not only an essential physiologic process required for normal development and maintenance of liver homeostasis, but is also involved in pathologic conditions, including liver regression, physical and chemical liver injury, viral hepatitis, and liver carcinogenesis.35
The aspartate-specific-cyteine-protease (caspase) cascade is now believed to be the main pathway by which cellular death is orchestrated.36 The most prevalent caspase in the cell is caspase-3. This caspase is ultimately responsible for the majority of the effects, and it is often referred to as an executioner caspase because of its role in coordinating the death of the cell. Upstream caspases such as caspases-8 and-9, are referred to as initiator caspases, indicating their role in triggering apoptosis by activating the executioners. There are two pathways by which caspase activation is triggered: the extrinsic and the intrinsic. The extrinsic pathway is activated by the engagement of death receptors on the cell surface.37 Caspase-8 is the key initiator caspase in the death-receptor pathway.38 The intrinsic pathway or non-receptor-mediated apoptosis is triggered by various extracellular and intracellular stresses, such as growth factor withdrawal, hypoxia, DNA damage and oncogene induction. Signals that are transduced in response to these stresses converge mainly on the mitochondria, and result in the permeabilization of the outer mitochondrial membrane, release of cytochrome c and other pro-apoptotic molecules, formation of the apoptosome and caspase activation.39 Among these processes, only the permeabilization step is regulated, and several members of the Bcl-2 family are involved in this regulation. The anti-apoptotic proteins Bcl-2 and Bcl-xL can stop the march towards apoptotic death by preventing cytochrome c release,40 whereas translocation of pro-apoptotic proteins (Bax, Bid) to the mitochondria can induce the release of cytochrome c contained in the intermembrane space. Once cytochrome c is released, the downstream cascade of caspase activation is irreversible.41
Although apoptosis may be initiated in any phase of the cell cycle, the majority of cells undergo apoptosis primarily in the G1 phase of cycling cells, and there is a positive relationship between apoptosis and proliferation of cells. This relationship is explained by the presence of many cell cycle regulators /apoptosis inducers such as p53, which operate at the G1/S checkpoint.
As regards to the effects of IFN-α on the cell cycle of various normal and tumor cell lines, most studies have observed inhibitory effects on Gj to S-phase transition;42 other studies revealed S-phase accumulation in response to IFN-α treatment.43 In our first study, the animals treated with IFN-α2b showed a diminution on the percentage of preneoplastic hepatocytes in S phase, and an accumulation in G1 phase. In this connection, we examined whether p53 and three members of the Bcl-2 family (Bax, Bcl-2 and Bcl-xL), which are important regulators of apoptosis, were involved in IFN-α2b-mediated apoptosis. It is known that p53 down regulates Bcl-244 and up regulates Bax genes.45 The role of the Bcl-2 family in IFN-α-induced apoptosis still remains controversial. For example, IFN-α-induced apoptosis in cells of hematopoietic and hepatic origins can occur without involvement of the Bcl-2 family46 whereas transfection of IFN-α-sensitive cell lines with a Bcl-2 expression vector conferred partial resistance to cell death mediated by IFN-α.47
Our results suggested that IFN-α2b treatment increased the levels of the proapoptotic protein Bax, in parallel with increases of p53 protein levels. Besides, there were decreases in the levels of Bcl-2 and Bcl-xL proteins, which are known that promote cell survival. The relative prevalence of Bax and Bcl-xL protein are critical factors influencing cell fate, promoting either survival or death, whose ultimate outcome largely depends on the Bax/Bcl-xL ratio. Thus, apoptosis pathways can be activated under conditions in which Bax protein expression is elevated and/or Bcl-xL protein expression is decreased.
We also observed increased Bax protein translocation into the mitochondria in the animals that received IFN-α2b. It has been established that subcellular localization of Bax protein is an important regulator of apoptosis. Bax is localized in the cytoplasm and translocates to the mitochondria at the early stage of apoptosis. Bax mediates its proapoptotic effects through a channel forming activity of the mitochondrial membrane (a sudden increase in permeability of mitochondrial membrane, the so-called mitochondrial permeability transition pore: MPTP), resulting in disruption of mitochondrial function, release of cyto-chrome c, and apoptosis.48
Our observations suggested that preneoplastic hepatocytes in the IFN-α2b-treated rats were “primed” for apoptosis, and underwent programmed cell death as a primary result of a substantial increase in the level of mitochondrial Bax protein producing a further increase in the Bax/Bcl-xL protein ratio.
Transforming growth factor ßTransforming growth factor ß-ligands TGFßp TGFß2 and TGFß3 elicit a broad range of cellular responses, including the regulation of cell growth, differentiation, matrix production, and apoptosis. Among these, growth inhibition by TGFβ for epithelial cells, endothelial cells, and hematopoietic cells has been of central interest because it may be instrumental in preventing malignant conversion of cells in the body.49 Indeed, tumor cells of diverse tissue origins lose their sensitivity to TGFβ-induced growth inhibition during the steps of malignant transformation.50
Despite the amazingly diverse set of cellular responses regulated by TGFβ, the central signaling pathway downstream of TGFβ is surprisingly simple. TGFβ exerts its various effects via two transmembrane serine/threonine kinases known as type I and type II receptors (TBRI and TBRII). The ligand-activated type II receptor associates with, phosphorylates, and activates the type I receptor, which in turn phosphorylates pathway-specific Smads-2 and-3, members of the Smad family of signal transducers.51 These activated Smads then associate with Smad-4 and translocate to the nucleus, where they regulate transcription by associating with nuclear transcription factors and/or by binding directly to DNA. The inhibitory Smads-6 and-7 bind stably TGFβ receptors and interfere with ligand-induced phosphorylation of Smads-2 and-3.52
TGFβ1 is an important physiological mediator of apoptosis in the liver. It is known that TGFβ1 induces apoptosis in primary hepatocyte cultures derived from both adult and fetal rat liver;53 intravenous administration of TGFβ1 induces apoptosis in both normal and regressing liver54 and also in hepatocytes from preneoplastic foci. TGFβ1 protein is expressed in apoptotic hepatocytes from both normal and preneoplastic liver.55 In addition, several hepatoma cell lines are sensitive toward programmed cell death induction by TGF/β1.56-57
The exact mechanism by which TGFβ1 mediates apoptosis is not completely understood. Induction of proapoptotic genes such as p53 and Bax as well as activation of caspase-8 has been shown for liver epithelial cells. Furthermore, high apoptotic death of liver cells from c-myc transgenic mice is accompanied by up-regulation of proapoptotic gene products, such as p53 and Bax and decreased Bcl-2 expression. TGFβ1, in agreement with its apoptosis-inducing activity, decreased the antiapoptotic protein Bcl-xL in diverse hepatoma cell lines. On the other hand, overexpression of Bcl-2 blocked induction of apoptosis by TGFβ1 in 2 human hepatoma cell lines.58
Nonparenchymal cells, including Kupffer cells and peritoneal macrophages (Mϕs), are the main source of hepatic TGFβ1 in normal liver,59 whereas hepatocytes produce any of the isoforms of TGFβ. In addition, Kupffer cell-secreted TGFβ1 plays a pivotal role in the pathogenesis of alcoholic and fibrotic liver diseases. Moreover, hepatocyte apoptosis in severe acute pancreatitis occurs via TGFβ1 derived from peritoneal Mϕs. Unlike original concepts, evidence suggests that hepatocytes may synthesize TGFβ1in vitro as well as during hepatocarcinogenesis. Finally, it was reported that IFN-α2b treatment exerted a significantly elevated TGFβ1 secretion in single Mϕs.60
Due to the data obtained in our previous work and those obtained by other authors, we studied if the soluble mediator TGFβ1 was responsible for the observed apoptosis of preneoplastic hepatocytes induced by IFN-α2b, and also we tried to clarify the source of TGFβ1 in IFN-α2b-treated rats.61
We observed that serum TGFβ1 levels in the animals given IFN-α2b were significantly increased. The number of TGFβ1-positive hepatocytes was also augmented. In accordance with this, we observed increased phosphorylation and nuclear translocation Smads-2/3 proteins, indicating activation of the TGFβ1 signaling pathway in IFN-α2b-treated rats.
On the other hand, peritoneal Mϕs, Kupffer cells, and hepatocytes from rats with preneoplasia were isolated and cultured with or without IFN-α2b, in order to know the source of TGFβ1. Neither peritoneal Mϕs nor Kupffer cells secreted detectable levels of TGFβ1 when they were stimulated with IFN-α2b. However, IFN-α2b presence in the culture media of hepatocytes induced several fold increases of TGFβ1 production. Moreover, IFN-α2b-stimulated cultured hepatocytes from preneoplastic livers showed elevated apoptosis, measured by fluorescence microscopy and caspase-3 activity. They presented higher nuclear accumulation of phosphorylated Smads-2/3, indicating increased TGFβ1 signaling. When anti-TGFβ1 was added to the culture media, TGFβ1 activation and apoptosis induced by IFN-α2b were blocked.
Taken together, these data clearly showed that TGFβ1, which is produced and secreted by hepatocytes from pre-neoplastic liver under IFN-α2b treatment, stimulated their apoptotic cell death in an autocrine/paracrine fashion. This postulated mode of action was in good agreement with data published previously.62,63 The reduction of preneoplastic foci by endogenous TGFβ1 early in the carcinogenesis process would likewise protect from tumor formation.
In summary, we demonstrated for the first time that the apoptotic effect of IFN-α2b on preneoplastic liver foci is mediated by the production of endogenous TGFβ1 from hepatocytes acting by a paracrine/autocrine way.
In order to go deeper into the mechanisms of the relationship between IFN-α2b and TGFβ1 in our preneoplastic rat liver model, we demonstrated, in another set of in vitro experiments, that endogenous TGFβ1 secreted under IFN-α2b stimulus seems to induce cytochrome c release through a mechanism related to Bcl-2 family members and induction of oxidative stress, with an increase in reactive oxygen species (ROS) and loss of mitochondrial transmembrane potential (▵Ψ). Bax protein could be responsible of the release of cytochrome c during the initial hours of IFN-α2b-induced apoptosis via TGFβ1, whereas at later times (after 20 h of culture) activation of Bid by caspases could amplificate the mitochondrial events, enhancing the release of cytochrome c (Alvarez et al. Manuscript submitted).
We also demonstrated in vitro (Quiroga et al. Manuscript submitted)) that IFN-α2b induced an early activation of NADPH oxidase enzyme complex in hepatocytes obtained from rat preneoplastic liver. Results lead us to conclude that IFN-α2b induces the early ROS production that serves as a messenger, promoting the TGFβ1 production and secretion. This growth factor triggers the production of more reactive oxygen intermediates, as a late event, by inducing the same enzyme complex, showing an additive response in ROS production and imposing the final onset of the apoptotic effect.
Besides, we found that the biosynthetic glutathione (GSH) capacity was altered and the activities of antioxidants enzymes (catalase (CAT), cytosolic and mitochondrial superoxide dismutase (SOD)) were decreased.
The presence of ascorbic acid (ASC) in the culture media totally blocked the increase in the activity of the NADPH oxidase complex at all the studied times. These results were in agreement with other authors that demonstrated that the activity of this complex is lowered by the presence of antioxidants such as ASC or vitamin E in several cell types.64,65
These results confirmed that the perturbation of the redox status produced by the IFN-α2b induction of NADPH oxidase complex triggered TGFβ1 synthesis and secretion and assess the downregulation of the antioxidative systems. Similar data have been reported by Herrera et al. when they treated fetal rat hepatocytes with TGFβ1.52
Since ASC abolished all the apoptotic effects induced in vitro by IFN-α2b, we determined the relevance of ROS on the onset of the apoptotic process in vivo in the whole preneoplastic liver. Treatment of preneoplastic rats with IFN-α2b + ASC abolished the IFN-α2b apoptotic effects observed in IFN-α2b-treated rats.
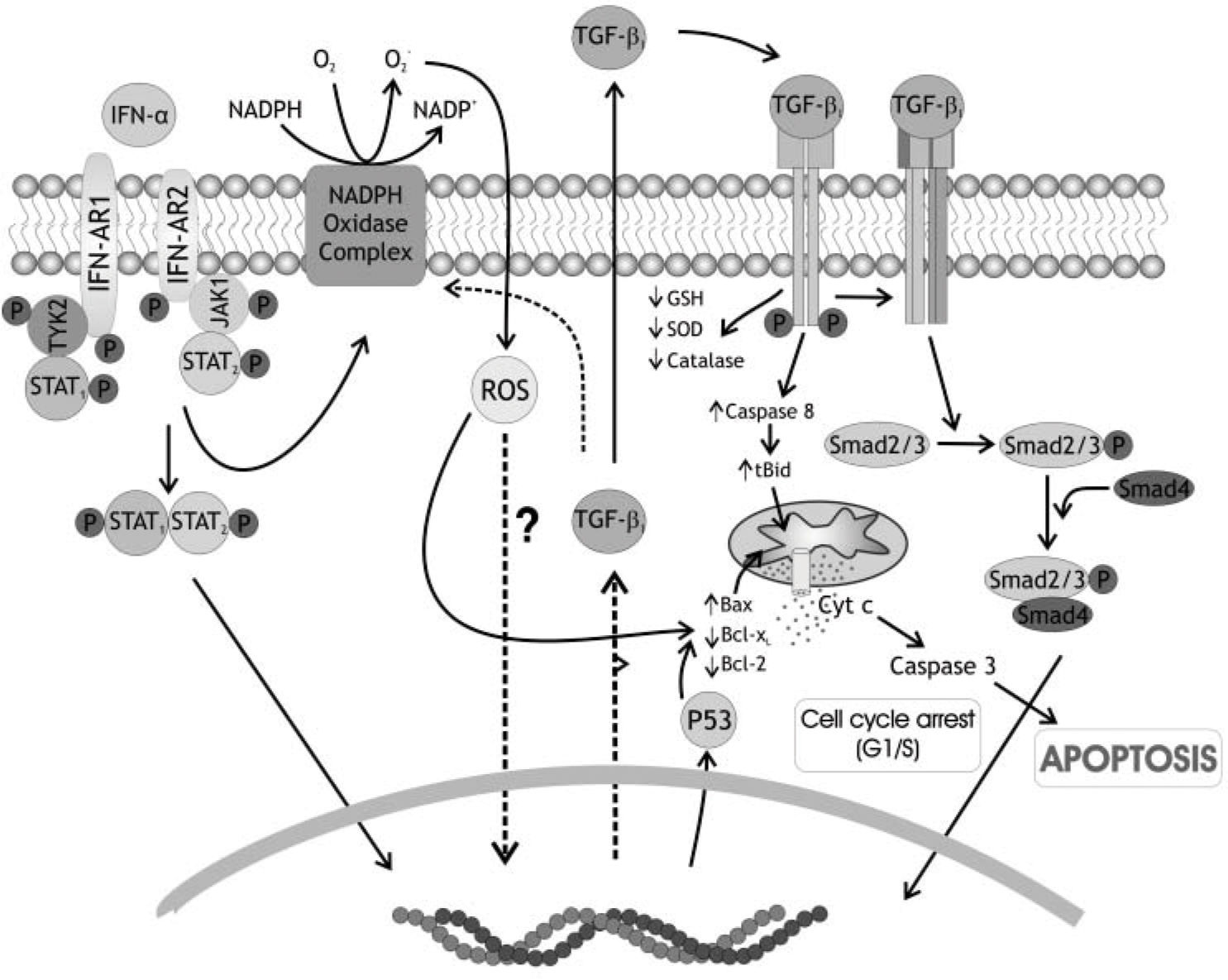
At the light of previous knowledge and from our own results we postulate the following events sequence to explain the IFN-α2b action on hepatocytes of preneoplastic liver (Figure 1):
. ROS, in turn, trigger TGF ß1 production and secretion. TGF ß when binds to its receptor also induces NADPH oxidase complex, and, besides, decreases the antioxidant defenses of the cell: glutathione content (GSH), catalase activity (CAT), superoxide dismutase activity (SOD). ROS initiate mitochondrial apoptosis directly and/or acting by the Bcl-2 family proteins inducing a mitochondrial permeability transition pore (MPTP), releasing of cytochrome c and the activation of caspase 3. TGF ß1 could induce, as a late event, the activation of caspase 8, which, in turn, induces a higher MPTP through activation of Bid, another Bcl-2 family member.")
Schematic representation of IFN-α2b action on hepatocytes of preneoplastic rat livers. NADPH oxidase complex is activated by IFN-α2b binding to type I receptor. This binding produces early amounts of reactive oxygen species (ROS). ROS, in turn, trigger TGF ß1 production and secretion. TGF ß when binds to its receptor also induces NADPH oxidase complex, and, besides, decreases the antioxidant defenses of the cell: glutathione content (GSH), catalase activity (CAT), superoxide dismutase activity (SOD). ROS initiate mitochondrial apoptosis directly and/or acting by the Bcl-2 family proteins inducing a mitochondrial permeability transition pore (MPTP), releasing of cytochrome c and the activation of caspase 3. TGF ß1 could induce, as a late event, the activation of caspase 8, which, in turn, induces a higher MPTP through activation of Bid, another Bcl-2 family member.
a) Binding of IFN-α2b to type I receptor in the surface of hepatocyte, b) activation of NADPH oxidase complex in hepatocyte membrane, c) production of ROS, d) induction of TGFβ1 synthesis and secretion, e) binding of TGFβ1 newly synthetized to its receptor on the surface of hepatocyte, f) new induction of NADPH oxidase complex, g) diminution of antioxidants defenses, h) induction of p53 and Bax synthesis, i) translocation of Bax to mitochondria and production of MPTP, j) activation of caspase 8, k) activation of Bid, production of tBid and increase of MPTP, l) release of cytochrome c from mitochondria, m) activation of caspase 3 and apoptosis.
ConclusionsIn the context of using type I IFNs as therapeutics agents in the treatment of human liver diseases, the use of antioxidants could have the potential to decrease effectiveness of the therapy. Besides, the potential feed-back regulation or interference via other cytokines and growth factors deserve more consideration.
The complexity that is associated with the TGFβ1-mediated regulation of cell behaviour in early stages of cancer is further convoluted by the disease itself, as many networks are often misregulated and amplified in cancer cells to promote progression. It seems from our results that IFN-α2b could enhance the proapoptotic effects of TGFβ1 in early stages of hepatocarcinogenesis, which could be highly beneficial in cancer therapy.
Undoubtedly, some time from now a more profound understanding of the complex cytokine and growth factors network will emerge, and this will help in these issues.



