



Liquid biopsy, specifically the analysis of circulating tumor DNA (ctDNA), offers a non-invasive approach for hepatocellular carcinoma (HCC) diagnosis and management. However, its implementation in the clinical setting is difficult due to challenges such as low ctDNA yield and difficulty in understanding the mutation signals from background noise. This review highlights the crucial role of artificial intelligence (AI) in addressing these limitations and in improving discoveries in the field of liquid biopsy for HCC care. Combining AI with liquid biopsy data can offer a promising future for the discovery of novel biomarkers and an AI-powered clinical decision support system (CDSS) can turn liquid biopsy into an important tool for personalized management of HCC. Despite the current challenges, the integration of AI shows promise to significantly improve patient outcomes and revolutionize the field of oncology.
Liver cancer is the sixth most common cancer, and it is now the third leading cause of cancer-related death worldwide. In 2020, more than 900,000 new cases were diagnosed globally, and more than 800,000 liver cancer-related deaths were reported [1]. Hepatocellular carcinoma (HCC) represents about 75 %–85 % of primary liver cancers being a major health burden worldwide [2]. In Europe, the age-standardized mortality rate is around 3.8–4.0 per 100,000 population [2]. While in the US, the survival rates for HCC patients remain low with only a 3–34 % 5-year survival rate [3].
HCC can arise from various causes of chronic liver injury but regardless of etiology, it is suggested that a common mechanism leading to it is the continuous cycle of cell death and regeneration [4]. This can result in accumulation of multiple genetic events such as somatic mutations, copy number alterations, pathway alterations, and epigenetic changes that can lead to HCC initiation, progression, and metastasis [4]. Advancements in sequencing techniques allowed molecular characterization of tumors which can provide an understanding of the genomic landscape of HCC and help identify driver mutations [4–6]. This supports the rise of precision medicine in which there is a shift from a “one-size fits all” approach to an individualized management approach however, challenges are met in the collection and analysis of tumor tissue [6,7]. Archived tissues can be used to analyze mutations, but it may not contain enough tumor content to satisfy test sensitivity and may be degraded to provide any pertinent information [5]. It should also be highlighted that in the past, tissue biopsy for liver tumors was discouraged due to the so-called “seeding” problem. In addition, liver cancer exhibits significant intratumoral heterogeneity, making tissue biopsies less representative. In this regard, liquid biopsy offers great potential to overcome these limitations since it consists of a minimally-invasive analysis of biological fluids, most commonly blood, that can be used to detect cancer at early stages, thus giving patients good chances for effective treatment [8].
In recent years, a wide range of liquid-biopsy tests has been developed primarily using circulating tumor proteins, nucleic acids, such as circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), and tumor-derived extracellular vesicles (EVs). The use of liquid biopsy, particularly ctDNA analysis, has emerged as a promising approach in the field of liver cancer diagnostics and monitoring [8]. However, challenges such as low DNA yield [9,10] and the difficulty in deciphering mutation signals against background noise [11] have limited its widespread clinical implementation. In this minireview, we emphasize the crucial role of artificial intelligence (AI) in addressing these limitations and accelerating discoveries in the field of liquid biopsy for liver cancer.
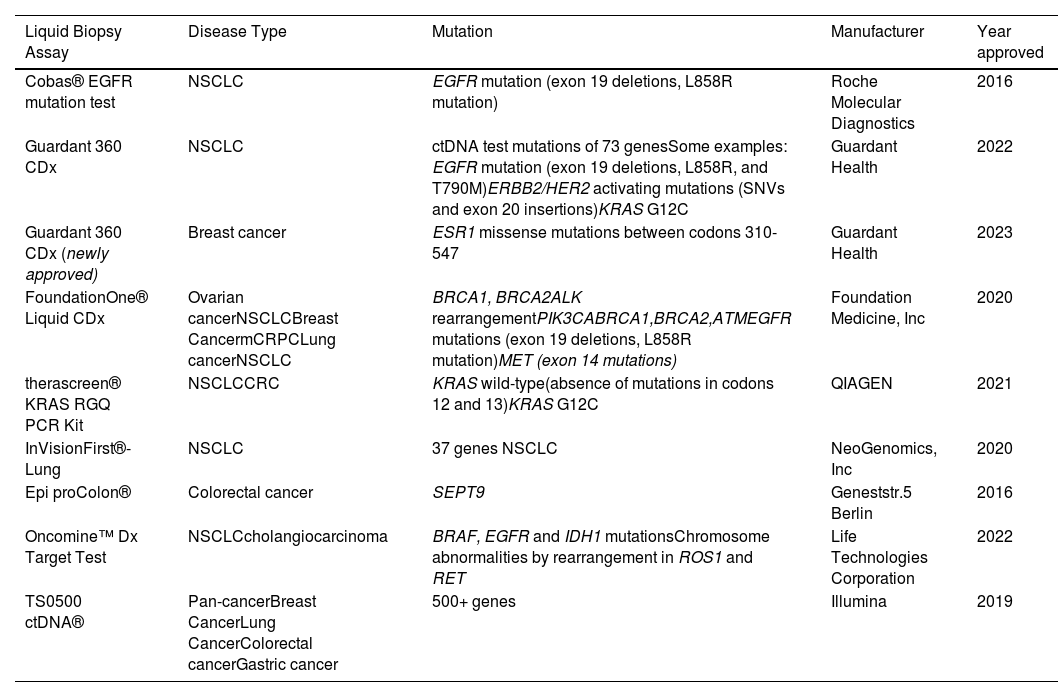
2Liquid biopsyInterest in liquid biopsy has continued to grow over the years and has led to approval for blood-based tests for precision cancer care [12]. In 2013, the US Food and Drug Administration (FDA) approved the CellSearchTM CTC enumeration platform, which enumerates epithelial circulating tumor cells (CTCs) (Table 1) [12]. This groundbreaking approval paved the way for the implementation of liquid biopsy in clinical trials such as the METABREAST trial, where CTC counts were used as a criterion for selecting first-line treatment in metastatic breast cancer (Table 1) [8,13].
FDA approved liquid biopsy tests.
| Liquid Biopsy Assay | Disease Type | Mutation | Manufacturer | Year approved |
|---|---|---|---|---|
| Cobas® EGFR mutation test | NSCLC | EGFR mutation (exon 19 deletions, L858R mutation) | Roche Molecular Diagnostics | 2016 |
| Guardant 360 CDx | NSCLC | ctDNA test mutations of 73 genesSome examples: EGFR mutation (exon 19 deletions, L858R, and T790M)ERBB2/HER2 activating mutations (SNVs and exon 20 insertions)KRAS G12C | Guardant Health | 2022 |
| Guardant 360 CDx (newly approved) | Breast cancer | ESR1 missense mutations between codons 310-547 | Guardant Health | 2023 |
| FoundationOne® Liquid CDx | Ovarian cancerNSCLCBreast CancermCRPCLung cancerNSCLC | BRCA1, BRCA2ALK rearrangementPIK3CABRCA1,BRCA2,ATMEGFR mutations (exon 19 deletions, L858R mutation)MET (exon 14 mutations) | Foundation Medicine, Inc | 2020 |
| therascreen® KRAS RGQ PCR Kit | NSCLCCRC | KRAS wild-type(absence of mutations in codons 12 and 13)KRAS G12C | QIAGEN | 2021 |
| InVisionFirst®-Lung | NSCLC | 37 genes NSCLC | NeoGenomics, Inc | 2020 |
| Epi proColon® | Colorectal cancer | SEPT9 | Geneststr.5 Berlin | 2016 |
| Oncomine™ Dx Target Test | NSCLCcholangiocarcinoma | BRAF, EGFR and IDH1 mutationsChromosome abnormalities by rearrangement in ROS1 and RET | Life Technologies Corporation | 2022 |
| TS0500 ctDNA® | Pan-cancerBreast CancerLung CancerColorectal cancerGastric cancer | 500+ genes | Illumina | 2019 |
Subsequently, in 2016, the first ctDNA-based companion diagnostic test, known as the Cobas EGFR Mutation Test v2, was developed to detect epidermal growth factor receptor (EGFR)-sensitizing mutations in non-small cell lung cancer (NSCLC) patients (Table 1). This test played a crucial role in guiding the use of EGFR-tyrosine kinase inhibitors as a targeted therapy [14]. With the rise of next-generation sequencing techniques, multigene panels were developed to detect target mutations in several cancer-related genes in advanced cancer. In 2019, FDA approved some liquid biopsy tests. One of these is the Guardant360 CDx (Table 1), which analyzes a 73-gene panel of cell-free DNA (cfDNA) to guide treatment for patients with NSCLC. Another is CancerSEEK, which identifies eight prevalent cancer types through the analysis of eight protein biomarkers combined with tumor-specific mutations in cfDNA of cancer patients [12]. Additional FDA-approved liquid biopsy tests are outlined in Table 1. These examples highlight how these tests have improved the selection of patients who could derive greater benefit from targeted therapies, signifying a noteworthy advancement in personalized medicine within the field of oncology.
HCC in particular presents considerable challenges in its management and treatment due to pronounced tumor heterogeneity, both in terms of intra- and inter-tumor molecular diversity [15]. Multinodular HCCs, diagnosed in approximately 41 % to 75 % of patients, limit the availability of curative treatment options and often lead to unfavorable prognosis [16]. The extensive tumor heterogeneity, particularly in multicentric HCC cases, could be underestimated when assessed through a single tumor biopsy, posing significant obstacles to precision medicine and the development of biomarker-based solutions in HCC [16,17].
Advancements in liquid biopsy, particularly the analysis of ctDNA, have revolutionized oncology by enhancing early detection and improving monitoring and targeted treatments through the evaluation of genomic and molecular profiles of tumors [17]. In contrast to various other cancers (as shown in Table 1), the field of liquid biopsy for HCC has been characterized by a lack of extensive and comprehensive studies able to provide sufficient and consistent results. This dearth of robust data has contributed to challenges in attaining FDA approvals for HCC-related applications.
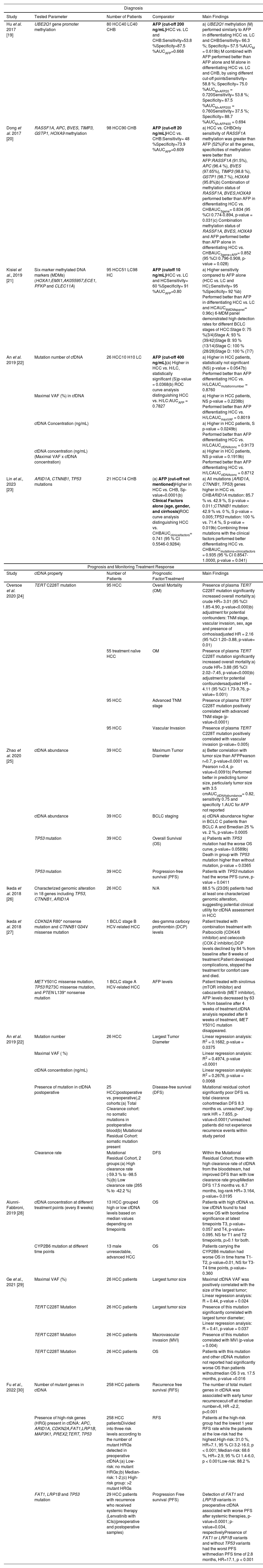
3Circulating tumor DNA in HCCcfDNA, first reported in human peripheral blood in 1948 by Mandel and Metais, exists as double-stranded fragments of approximately 150 to 200 base pairs in length. In healthy individuals, cfDNA derived from apoptotic myeloid and lymphoid cells is present at low levels (about 10 to 15 ng per milliliter) [18]. However, in the presence of tumors, inflammation, tissue damage, or after surgery, the concentration of cfDNA can increase in the bloodstream. ctDNA, a subset of cfDNA, refers specifically to fragmented DNA originating from tumor cells and it accounts for a fluctuating proportion of cfDNA, ranging from less than 0.1 % to over 90 % [9]. Generally, cfDNA levels are elevated in patients with carcinoma compared to healthy individuals. Significant amounts of ctDNA are released into the circulatory system through tumor cell apoptosis or necrosis, and its quantity can reflect the tumor burden in patients with cancer [9]. Table 2 provides a comprehensive overview of some studies exploring the potential of liquid biopsy tests for diagnosis, prognosis and treatment monitoring of HCC.
Liquid biopsy studies based on ctDNA analysis in HCC.
| Diagnosis | ||||
|---|---|---|---|---|
| Study | Tested Parameter | Number of Patients | Comparator | Main Findings |
| Hu et al. 2017 [19] | UBE2Q1 gene promoter methylation | 80 HCC40 LC40 CHB | AFP (cut-off 200 ng/mL)HCC vs. LC and CHB:Sensitivity=53.8 %Specificity=87.5 %AUCAFP=0.668 | a) UBE2Q1 methylation (M) performed similarly to AFP in differentiating HCC vs. LC and CHBSensitivity= 66.3 %; Specificity= 57.5 %AUCM = 0.619b) M combined with AFP performed better than AFP alone and M alone in differentiating HCC vs. LC and CHB, by using different cut-off pointsSensitivity= 58.8 %; Specificity= 75.0 %AUCM+AFP20 = 0.720Sensitivity= 53.8 %; Specificity= 87.5 %AUCM+AFP200 = 0.760Sensitivity= 37.5 %; Specificity= 88.7 %AUCM+AFP400 = 0.694 |
| Dong et al. 2017 [20] | RASSF1A, APC, BVES, TIMP3, GSTP1, HOXA9 methylation | 98 HCC90 CHB | AFP (cut-off 20 ng/mL)HCC vs. CHB:Sensitivity= 48 %Specificity=73.9 %AUCAFP=0.609 | a) HCC vs. CHBOnly sensitivity of RASSF1A methylation was greater than AFP (52%)For all the genes, specificities of methylation were better than AFP:RASSF1A (91.5%), APC (96.4 %), BVES (97.65%), TIMP3 (98.8 %), GSTP1 (98.7 %), HOXA9 (95.8%)b) Combination of methylation status of RASSF1A, BVES,HOXA9 performed better than AFP in differentiating HCC vs. CHBAUC3gene= 0.834 (95 %CI 0.774-0.894, p-value = 0.031)c) Combination methylation status of RASSF1A, BVES, HOXA9 and AFP performed better than AFP alone in differentiating HCC vs. CHBAUC3gene+AFP= 0.852 (95 %CI 0.796-0.908, p-value = 0.028) |
| Kisiel et al., 2019 [21] | Six-marker methylated DNA markers (MDMs)(HOXA1,EMX1,AK055957,ECE1, PFKP and CLEC11A) | 95 HCC51 LC98 HC | AFP (cutoff 10 ng/mL)HCC vs. LC and HCSensitivity= 60 %Specificity= 91 %AUCAFP=0.80 | a) Higher sensitivity compared to AFP alone (HCC vs. LC and HC):Sensitivity= 95 %Specificity= 92 %b) Performed better than AFP in differentiating HCC vs. LC and HCAUC6MDMspanel= 0.96c) 6-MDM panel demonstrated high detection rates for different BCLC stages of HCC:Stage 0: 75 %(3/4)Stage A: 93 % (39/42)Stage B: 93 % (13/14)Stage C: 100 % (28/28)Stage D: 100 % (7/7) |
| An et al. 2019 [22] | Mutation number of cfDNA | 26 HCC10 H10 LC | AFP (cut-off 400 ng/mL)(a) Higher in HCC vs. H/LC, statistically significant (S)p-value = 0.0368(b) ROC curve analysis distinguishing HCC vs. H/LC:AUCAFP = 0.7827 | a) Higher in HCC patients, statistically not significant (NS) p-value = 0.0547b) Performed better than AFP differentiating HCC vs. H/LCAUCmutationnumber = 0.8760 |
| Maximal VAF (%) in cfDNA | a) Higher in HCC patients, NS p-value = 0.2238b) Performed better than AFP differentiating HCC vs. H/LCAUCmaxVAF = 0.8019 | |||
| cfDNA Concentration (ng/mL) | a) Higher in HCC patients, S p-value = 0.0249b) Performed better than AFP differentiating HCC vs. H/LCAUCcfDNAconc = 0.9173 | |||
| ctDNA concentration (ng/mL) (Maximal VAF x cfDNA concentration) | a) Higher in HCC patients, NS p-value = 0.1919b) Performed better than AFP differentiating HCC vs. H/LCAUCctDNAconc = 0.8712 | |||
| Lin et al., 2023 [23] | ARID1A, CTNNB1, TP53 mutations | 21 HCC14 CHB | (a) AFP (cut-off not mentioned)Higher in HCC vs. CHB, Sp-value=0.0001(b) Clinical Factors alone (age, gender, and cirrhosis)ROC curve analysis distinguishing HCC vs. CHBAUCclinicalfactors= 0.741 (95 % CI 0.5546-0.9284) | a) All mutations (ARID1A, CTNNB1, TP53) genes higher in HCC vs. CHBARID1A mutation: 85.7 % vs. 42.9 %, S p-value = 0.011;CTNNB1 mutation: 42.9 % vs. 0 %, S p-value = 0.005;TP53 mutation: 100 % vs. 71.4 %, S p-value = 0.019b) Combining three mutations with the clinical factors performed better differentiating HCC vs. CHBAUCmutations+clinicalfactors = 0.935 (95 % CI 0.8547-1.0000, p-value = 0.041) |
| Prognosis and Monitoring Treatment Response | ||||
| Study | ctDNA property | Number of Patients | Prognostic Factor/Treatment | Main Findings |
| Oversoe et al. 2020 [24] | TERT C228T mutation | 95 HCC | Overall Mortality (OM) | Presence of plasma TERT C228T mutation significantly increased overall mortality:a) crude HR= 3.01 (95 %CI 1.85-4.90, p-value<0.000)b) adjustment for potential confounders: TNM stage, vascular invasion, sex, age and presence of cirrhosisadjusted HR = 2.16 (95 %CI 1.20–3.88, p-value= 0.01) |
| 55 treatment naïve HCC | OM | Presence of plasma TERT C228T mutation significantly increased overall mortality:a) crude HR= 3.88 (95 %CI 2.02–7.45, p-value<0.000)b) adjustment for potential confoundersadjusted HR = 4.11 (95 %CI 1.73-9.76, p-value= 0.001) | ||
| 95 HCC | Advanced TNM stage | Presence of plasma TERT C228T mutation positively correlated with advanced TNM stage (p-value<0.0001) | ||
| 95 HCC | Vascular Invasion | Presence of plasma TERT C228T mutation positively correlated with vascular invasion (p-value= 0.005) | ||
| Zhao et al. 2020 [25] | ctDNA abundance | 39 HCC | Maximum Tumor Diameter | a) Better correlation with tumor size than AFPPearson r=0.7, p-value<0.0001 vs. Pearson r=0.4, p-value=0.0091b) Performed better in predicting tumor size, particularly tumor size with 3.5 cmAUCctDNAabundance= 0.82, sensitivity 0.75 and specificity 1.AUC for AFP not reported |
| ctDNA abundance | 39 HCC | BCLC staging | a) ctDNA abundance higher in BCLC C patients than BCLC A and Bmedian 25 % vs. 2 %, p-value= 0.0005 | |
| TP53 mutation | 39 HCC | Overall Survival (OS) | a) Patients with TP53 mutation had the worse OS curve, p-value= 0.0589b) Death in group with TP53 mutation higher than without mutation, p-value = 0.0365 | |
| TP53 mutation | 39 HCC | Progression-free survival (PFS) | Patients with TP53 mutation had the worse PFS curve, p-value = 0.0411 | |
| Ikeda et al. 2018 [26] | Characterized genomic alteration in 18 genes including TP53, CTNNB1, ARID1A | 26 HCC | N/A | 88.5 % (23/26) patients had at least one characterized genomic alteration, suggesting potential clinical utility for ctDNA assessment in HCC |
| Ikeda et al. 2018 [27] | CDKN2A R80* nonsense mutation and CTNNB1 G34V missense mutation | 1 BCLC stage B HCV-related HCC | des-gamma carboxy prothrombin (DCP) levels | Patient treated with combination treatment with Palbociclib (CDK4/6 inhibitor) and celecoxib (COX-2 inhibitor).DCP levels declined by 84 % from baseline after 8 weeks of treatment.Patient developed complications, stopped the treatment for comfort care and died. |
| MET Y501C missense mutation, TP53 R273C missense mutation, and PTEN L139* nonsense mutation | 1 BCLC stage A HCV-related HCC | AFP levels | Patient treated with sirolimus (mTOR inhibitor) and cabozantinib (MET inhibitor), AFP levels decreased by 63 % from baseline after 4 weeks of treatment.ctDNA analysis repeated after 8 weeks of treatment, MET Y501C mutation disappeared. | |
| An et al. 2019 [22] | Mutation number | 26 HCC | Largest Tumor Diameter | Linear regression analysis: R2 = 0.1682, p-value = 0.0375 |
| Maximal VAF ( %) | Linear regression analysis: R2 = 0.4974, p-value <0.0001 | |||
| ctDNA concentration (ng/mL) | Linear regression analysis: R2 = 0.2676, p-value = 0.0068 | |||
| Presence of mutation in ctDNA postoperative | 25 HCC(postoperative vs. preoperative),2 cohorts:(a) Total Clearance cohort: no somatic mutations in postoperative blood(b) Mutational Residual Cohort: somatic mutation present | Disease-free survival (DFS) | Mutational residual cohort significantly poor DFS vs. total clearance cohortmedian DFS 8.3 months vs. unreached*, log-rank HR = 7.655, p-value<0.0001)*unreached: patients did not experience recurrence events within study period | |
| Clearance rate | Mutational Residual Cohort, 2 groups:(a) High clearance rate (-59.3 % to -98.5 %)(b) Low clearance rate (265 % to -42.2 %) | DFS | Within the Mutational Residual Cohort, those with high clearance rate of ctDNA from the bloodstream, had improved DFS than with low clearance rate groupMedian DFS 17.5 months vs. 6.7 months, log-rank HR= 3.164, p-value= 0.0195 | |
| Alunni-Fabbroni, 2019 [28] | cfDNA concentration at different treatment points (every 8 weeks) | 13 HCC grouped high or low cfDNA levels based on median values depending on timepoints | OS | Patients with high cfDNA vs. low cfDNA found to had worse OS with borderline significance at latest timepoints T3, p-value= 0.057 and T4, p-value= 0.095. NS for T1 and T2 timepoints, p>0.1 for both. |
| CYP2B6 mutation at different time points | 13 male unresectable, advanced HCC | OS | Patients carrying the CYP2B6 mutation had worse OS in time frame T1-T2, p-value=0.01, NS for T3-T4 time points, p-value= 0.360 | |
| Ge et al., 2021 [29] | Maximal VAF (%) | 26 HCC patients | Largest tumor size | Maximal ctDNA VAF was positively correlated with the size of the largest tumor; Linear regression analysis: R = 0.44, p-value = 0.024 |
| TERT C228T Mutation | 26 HCC patients | Largest tumor size | Presence of this mutation significantly correlated with largest tumor diameter; Linear regression analysis: R = 0.41, p-value = 0.037 | |
| TERT C228T Mutation | 26 HCC patients | Macrovascular invasion (MVI) | Presence of this mutation correlated with MVI (p-value = 0.004) | |
| TERT C228T Mutation | 26 HCC patients | OS | Patients with this mutation and other ctDNA mutation not reported had significantly worse OS than patients withoutmedian OS 3 vs. 17.5 months, p-value =0.016 | |
| Fu et al., 2022 [30] | Number of mutant genes in ctDNA | 258 HCC patients | Recurrence free survival (RFS) | The number of total mutant genes in ctDNA was associated with early tumor recurrencecut-off at median number=6, HR =2.2, p<0.001 |
| Presence of high-risk genes (HRG) present in ctDNA: APC, ARID1A, CDKN2A,FAT1,LRP1B, MAP3K1, PREX2,TERT, TP53 | 258 HCC patientsDivided into three risk levels according to the number of mutant HRGs detected in preoperative ctDNA:(a) Low-risk: no mutant HRGs;(b) Median-risk: 1-2;(c) High-risk group: >2 mutant HRGs | RFS | Patients at the high-risk group had the lowest 1 year RFS rate while the patients at the low-risk had the highest.High-risk: 31.0 %, HR=7.1, 95 % CI 3.2-16.0, p < 0.001; Median-risk: 68.6 %, HR= 2.9, 95 % CI 1.4-6.0, p < 0.001Low-risk: 88.2 % | |
| FAT1, LRP1B and TP53 mutation | 29 HCC patients with recurrence who received systemic therapy (Lenvatinib with ICIs)(preoperative and postoperative samples) | Progression Free survival (PFS) | Detection of FAT1 and LRP1B variants in preoperative ctDNA associated with worse PFS after systemic therapies, p-value<0.0001; p-value=0.034, respectivelyPresence of FAT1 or LRP1B variants and without TP53 variants had the worst PFS withmedian PFS time of 2.8 months, HR=17.1, p < 0.001 | |
HCC, hepatocellular carcinoma; LC, liver cirrhosis; CHB, chronic hepatitis B; M, UBE2Q1 methylation; H, hepatitis; HC, healthy controls; BL, Benign Lesions; NS, not statistically significant; S, statistically significant; ROC, receiver operating characteristic; AUC, area under the curve; MDM, methylated DNA markers; VAF, variant allele frequency; OM, overall mortality; OS, overall survival; PFS, progression-free survival; DFS, disease-free survival; MVI, macrovascular invasion; RFS, recurrence free survival.
cfDNA and ctDNA generally hold substantial diagnostic potential in HCC; both can provide heightened sensitivity and improved clinical correlation, enhancing their value as diagnostic tools [10]. The methylation profile of cfDNA is particularly intriguing, given that epigenetic modifications have been identified as significant contributors to tumor initiation and progression [3,31]. Several studies have reported that changes in methylation patterns of different genes can distinguish HCC from controls [19,21]. An example is the study by Dong et al. in 2017, in which they found that a combination of methylation of Ras association domain family 1A (RASSF1A), blood vessel epicardial substance (BVES) and homeobox (HOXA9) gene promoters in serum and AFP outperformed AFP alone in distinguishing between HCC and chronic hepatitis B patients without HCC (CHB). Indeed, the Area Under the Curve (AUC) increased from 0.609 to 0.852 (p-value=0.028) [20] (Table2). cfDNA and ctDNA can also harbor certain key driver mutations in common genes in HCC and their detection can serve as potential biomarkers for early detection of HCC [3]. In a study by Lin et al. in 2023, the mutations on three gene AT-rich interaction domain 1A (ARID1A), catenin beta 1 (CTNNB1), and tumor protein p53 (TP53) were detected in ctDNA. These mutations were found more prevalent in HCC patients compared to CHB patients (85.7 % vs. 42.9 %, p-value = 0.011; 42.9 % vs. 0 %, p-value = 0.005; 100 % vs. 71.4 %, p-value = 0.019, respectively). This study also demonstrated that combining the mutation profile of the three genes with clinical factors, such as age, gender, and presence of cirrhosis, improved the performance of the model for the detection of early-stage HCC in patients with HBV with the AUC increasing from 0.741 to 0.935 (p-value=0.041) [23] (Table 2).
In another study, An et al. in 2019 compared different cfDNA parameters such as mutation number, maximal variant allele frequency (VAF), cfDNA concentration and ctDNA concentration between non-HCC patients, specifically hepatitis/cirrhotic patients and HCC patients. They found cfDNA concentration (p-value =0.0249) was significantly higher in HCC patients compared to the controls. In addition, this study also showed that all the cfDNA parameters performed equally or better than AFP in differentiating between HCC and hepatitis/cirrhosis patients [22] (Table 2).
Hence, cfDNA and ctDNA hold a potential diagnostic value in HCC, showcasing high sensitivity and robust clinical relevance. The methylation profile of ctDNA and mutations within specific genes emerge as promising tools demonstrating improved results in HCC detection, opening new possibilities for better patient management.
3.2ctDNA to monitor HCC progression and treatment responseThe analysis of ctDNA finds multiple applications in the clinical management of HCC patients [29]. Oversoe et al. in 2020, detected the presence of telomerase reverse transcriptase (TERT) C228T mutation in the ctDNA derived from 44 % of the HCC patients enrolled in the study (n=96), while it was absent in patients with cirrhosis (n=45) [24]. After adjustment for potential confounders, the presence of TERT mutation in plasma was associated with a higher mortality rate (adjusted HR 2.16, 95 % CI 1.20-3.88, p=0.010). On the other hand, in treatment-naive HCC patients, the presence of TERT C228T mutation was strongly associated with mortality (adjusted HR 4.11, 95 % CI 1.73-9.76, p=0.001). Additionally, the researchers established a positive correlation between the presence of TERT mutation in plasma and advanced TNM stage (p-value<0.0001) or vascular invasion (p-value=0.005) [24] (Table 2). Thus, highlighting the possible role of TERT C228T mutation detected in ctDNA as a prognostic marker in HCC. ctDNA has been used as a reliable biomarker to monitor tumor progression and assess treatment efficacy [28,30]. Due to the invasiveness of tissue biopsy, this procedure is highly discouraged to monitor the tumor progression, thus liquid biopsy provides an ideal option for real-time monitoring of the disease. A prospective study conducted by Zhao et al. in 2020 enrolled 42 patients with unresectable liver cancer, 39 of which had HCC [25]. The primary objective of the study was to investigate the relationship between ctDNA abundance and tumor characteristics, as along with the significance of TP53 mutations on response to interventional therapy. The results of the study revealed a strong correlation between ctDNA abundance and tumor size, highlighting the potential of ctDNA as a potential marker for quantifying tumor burden [25]. In addition, increased ctDNA levels were associated with the BCLC staging, indicating its potential as a prognostic biomarker [25]. Furthermore, patients without TP53 mutations showed improved overall survival (OS) and progression-free survival (PFS) compared to patients with TP53 mutations (OS: p-value=0.0589; PFS: p-value=0.0411) (Table 2). These findings suggest a possible association of this mutation with a less favorable response to interventional therapy, but further research with larger sample size is needed to confirm the results [25].
Indeed, the incorporation of ctDNA analysis into clinical practice enhances precision medicine approaches and improves patient outcomes in the management of HCC. Another example is a recent program called the TARGET (Tumour chARacterisation to Guide Experimental Targeted Therapy) study that aimed to assess the feasibility of using ctDNA to identify actionable mutations in early-phase clinical trials in a range of advanced-stage cancers [32]. In the initial phase, the study enrolled 100 patients, and 41 had actionable alterations. Among them, 11 received matched molecular therapies that led to a stabilization of the disease or to a partial response in patients with advanced cancers [32]. Similarly in HCC, another study identified potentially actionable alterations in ctDNA of 26 patients with advanced HCC involving 18 genes with the most common genetic alterations found in TP53, CTNNB1, and ARID1A genes [26] (Table 2). In a separate study, two HCC patients with specific somatic alterations in ctDNA received targeted drugs corresponding to their mutations, resulting in a decline in tumor marker (Alpha-Fetoprotein (AFP) and des‐gamma‐carboxy prothrombin (DCP)) during treatment [27]. In particular, one patient harboring a cyclin-dependent kinase inhibitor 2A (CDKN2A)‐inactivating and a CTNNB1‐activating mutation, received matched treatments consisting of palbociclib, a cyclin-dependent kinase (CDK)4/6 inhibitor, and celecoxib, a cyclooxygenase (COX)‐2/WNT inhibitor. Within 2 months DCP levels were reduced by 84 % [27] (Table 2). Another patient harboring phosphatase and tensin homolog deleted on chromosome 10 (PTEN)‐inactivating and a mesenchymal-epithelial transition (MET)‐activating mutations was treated with sirolimus (mechanistic target of rapamycin (mTOR) inhibitor) and cabozantinib (MET inhibitor) [27]. Imaging conducted 6 weeks after treatment indicated stable disease with central necrosis evident within the tumors, accompanied by a reduction in AFP levels within a month. Subsequent ctDNA analysis performed after 8 weeks of cabozantinib treatment revealed the absence of the MET mutation (MET Y501C), aligning with a positive molecular response to therapy [27] (Table 2). These studies evidenced the feasibility of a liquid biopsy-driven targeted therapy that can improve patient outcome.
4Artificial intelligence can improve ctDNA-based liquid biopsy testsThus far, the accumulating evidence from the literature holds promise for the prospective application of liquid biopsy in clinical oncology.
However, it should be considered that ctDNA constitutes only a minor fraction of the total cfDNA present in the plasma of cancer patients [9,10] and in the case of early or very early cancer stage it is even less abundant. For example, data derived from different studies in NSCLCs estimated the clonal mutant allele frequencies (MAFs) of ctDNA in the total cfDNA ranging from 0.01 % to 9.3 % (median 0.31 %) in patients with stage I [33]. Additionally, the “background noise “ derived from white blood cells (WBC) makes the detection of the ctDNA mutations even more difficult and less reliable [11] possibly resulting in a misinterpretation of the results or a misclassification of the patient.
New technical and analytical approaches should be adopted to address these limitations. The availability of more sensitive techniques, such as ultra-deep sequencing, that enables the identification of target ctDNA at very low concentrations [31,20], opened new opportunities in this field. This technique became cost-benefit advantageous in the case of targeted sequencing, where specific amplicons and genes are analyzed to detect known or novel mutations. However, many cancers, including HCC, lack a set of known mutations that can identify a tumor. Thus, the fundamental step that will improve liquid biopsy tests based on ctDNA is the identification of a specific mutational panel for HCC. In this regard, Artificial Intelligence (AI) can offer great help to analyze and identify specific combinations of mutations that “label” specifically the cancer, possibly using data available in genomic databases [34].
Recently, several works demonstrated the benefits derived from the application of machine learning algorithms to liquid biopsy tests. For example, Roth et al. used the filter-based feature method and Support Vector Machine (SVM), a supervised learning method for solving data mining problems, to filter 1158 miRNAs collected from blood to determine a suitable subset of biomarkers in glioblastoma [35]. They achieved the best performance by combining 180 miRNAs with an accuracy of 81 %, specificity of 79 %, and sensitivity of 83 % [34,35]. Another example is the combination of Single strand Adaptor Library Preparation (SALP)-sequence and SVM in the pursuit of uncovering novel cfDNA-based biomarkers for esophageal cancer, as demonstrated by Liu et al.[36]. In this study, the analysis of 30 cfDNA samples derived from 26 esophageal cancer (ESCA) patients and 4 healthy individuals resulted in the identification of 103 epigenetic markers and 37 genetic markers [36], thus proving the advantage conferred by AI technologies within the context of liquid-based tests.
Enhancements can be further obtained by combining the information derived from ctDNA with additional multi-omics data, including, transcriptomic, and proteomic information, all sourced from a single blood sample. Here AI methodologies play a pivotal role in seamlessly integrating these diverse layers of molecular data, illuminating concealed associations among them. By combining AI algorithms with the reservoir of information derived from liquid biopsy, researchers can gain a deeper understanding of the underlying biology of liver cancer, identify novel biomarkers, and develop more effective treatment strategies. An example of this concept is the study conducted by Chen et al. wherein they introduced an innovative integrated approach called HIFI (5-Hydroxymethylcytosine/motIf/Fragmentation/nucleosome footprint) [37]. This method was developed to diagnose HCC by analyzing cfDNA genomic features. Impressively, HIFI outperformed the conventional AFP biomarker in terms of sensitivity and specificity. This novel method emerges as a promising non-invasive and accurate strategy for early HCC diagnosis and surveillance in high-risk populations, highlighting the importance of cfDNA analysis in cancer detection [37].
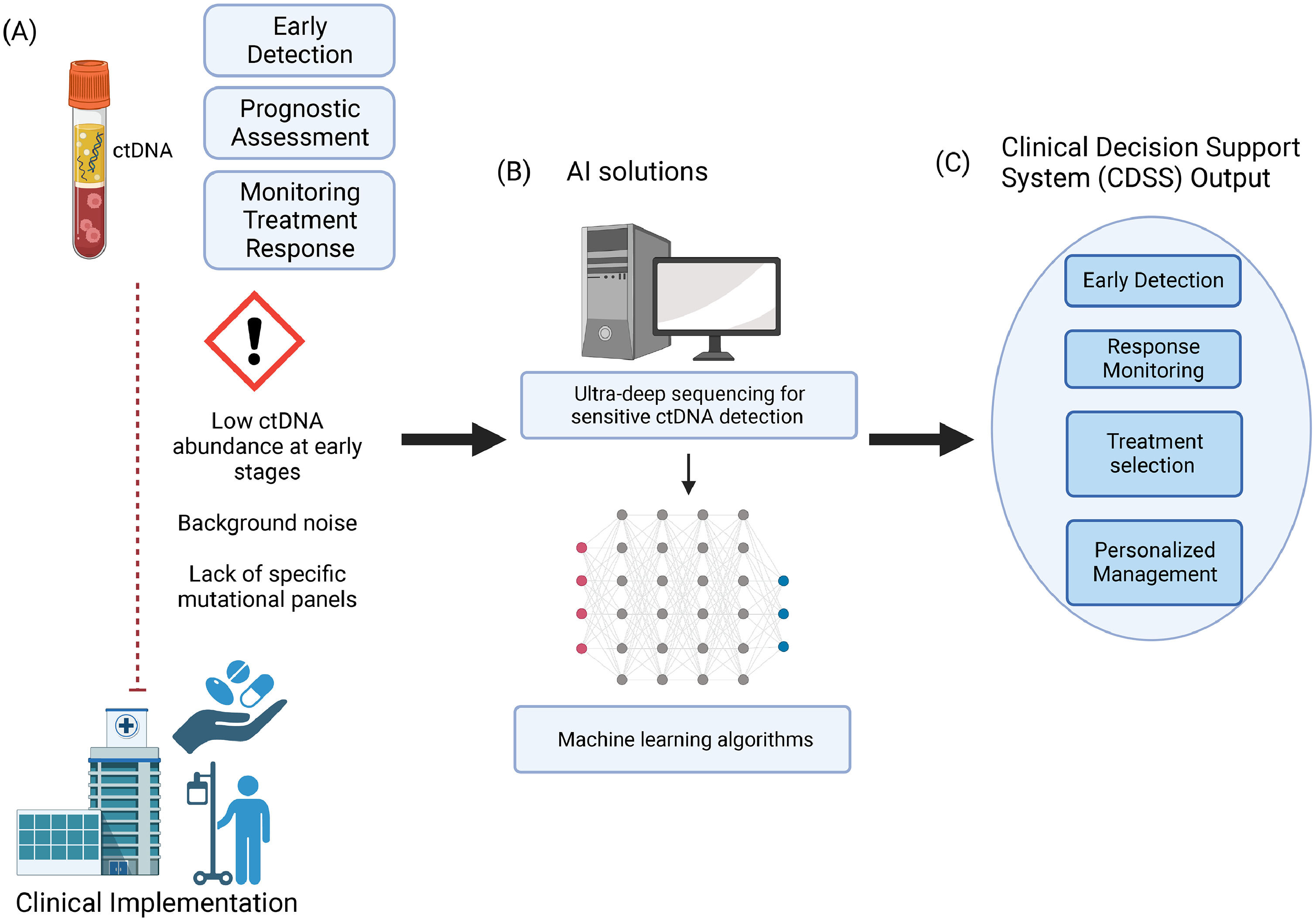
5Liquid biopsy and AI boosts the development of clinical decision support systemsAI-based clinical decision support systems (CDSS) have the potential to transform the field of liquid biopsy for liver cancer (Fig. 1). By analyzing large-scale patient data, including ctDNA profiles, treatment responses, and clinical outcomes, AI algorithms can generate predictive models and treatment recommendations [38]. CDSS powered by AI can aid clinicians in making informed decisions, selecting appropriate therapies, and monitoring treatment responses. Several groups have explored this potential and have reported promising results. An example is a study by Visser et al. which evaluated the performance of liquid biopsy-based decision support algorithms for the diagnosis and subtyping of lung cancer. They found that multi-parametric models combining assessment of protein tumor markers (cancer antigen 125 (CA125), cancer antigen 15-3 (CA15.3), carcinoembryonic antigen (CEA), cytokeratin 19 fragment 21-1 (CYFRA 21-1), human epididymis protein 4 (HE4), neuron-specific enolase (NSE), pro gastrin-releasing peptide (proGRP), and squamous cell carcinoma antigen (SCCA)) and ctDNA tumor markers (ctDNA mutations in epidermal growth factor receptor (EGFR), Kirsten rat sarcoma virus (KRAS) and b-raf proto-oncogene (BRAF)) were able to identify two-thirds of all lung cancer and NSCLC patients, as well as half of the small-cell lung cancer (SCLC) patients [39]. These findings underscore the specific significance of the models in scenarios where traditional lung tissue biopsies prove impractical or provide inconclusive outcomes. Furthermore, the study revealed the utility of baseline ctDNA measurements for monitoring treatment responses [39].
: Despite the potential of liquid biopsy, particularly ctDNA, still some limitations exist that limit its clinical use, (B): AI can play a crucial role in addressing these limitations, during the study design, during the analytical phase, and (C) in the interpretation of the results thought AI-powered clinical decision support systems (CDSS). CDSS can aid in early detection, treatment selection, and monitoring of the response to treatment leading to personalized management of the patient. The Figure was created on Biorender.com, licensed under Biorender")
Challenges in liquid biopsy tests and offered solutions by AI in Clinical Oncology. (A): Despite the potential of liquid biopsy, particularly ctDNA, still some limitations exist that limit its clinical use, (B): AI can play a crucial role in addressing these limitations, during the study design, during the analytical phase, and (C) in the interpretation of the results thought AI-powered clinical decision support systems (CDSS). CDSS can aid in early detection, treatment selection, and monitoring of the response to treatment leading to personalized management of the patient. The Figure was created on Biorender.com, licensed under Biorender's Academic License Terms. (https://www.biorender.com/academic-license)
Now, despite these potential clinical implications, there are still some discussion on how it can be optimally integrated in the clinical decision-making process [39]. The use of AI to analyze circulating biomarkers, such as cell-free DNA (cfDNA) for early detection has become increasingly important in clinical practice as this has been credited to reduce morbidity and mortality of cancer patients. Efforts to develop and validate multi-cancer early detection (MCED) tests were made by the Circulating Cell-free Genome Atlas (CCGA), a prospective, multicenter, case-control, observational study. CCGA is funded by GRAIL, Inc and involves collaboration with various researchers and institutions. In this study, participants with and without cancer are enrolled and cell-free DNA (cfDNA) sequencing is done in combination with machine learning to detect cancer signals across multiple cancer types and predict cancer signal origin (CSO) with high accuracy [40]. The CCGA was divided into three substudies which include a comprehensive comparison of genomic sequencing approaches, refinement of the selected assay and classifiers and a large clinical validation study of the MCED test [40].
`The first substudy focused on evaluating features related to cfDNA in experimental tests and prototype machine-learning classifiers [41]. It aimed to identify the most promising approach for a MCED test with a low false-positive rate and sufficient sensitivity to improve outcomes. The results showed that cfDNA methylation was the most promising genomic feature for cancer signal detection and the whole-genome methylation-based approach was selected for further development and improvement into a targeted methylation assay, and machine learning classifier for cancer detection and CSO prediction in the second substudy [41]. The third and last CCGA substudy reported on a large clinical validation study of this refined MCED test, GalleriTM MCED test. This test can detect more than 50 different cancer types and has shown high specificity for cancer signal detection (99.5 %) and accurate prediction of cancer signal origin with 4,077 study participants involving cancer and non-cancer individuals [40]. In addition, this test correctly predicted cancer tissue of origin in 88.7 % of true-positive cases. Validation in an independent set further confirmed its efficacy in detecting cancer signals [40]. These findings indicate its potential as a complementary screening tool for detecting multiple cancer types and improving clinical outcomes. Follow-up for all participants for clinical outcome of the CCGA study is still ongoing and the estimate completion is the first quarter of 2024 [42].
Several CDSS have already been developed and some are available in the market and have undergone thorough reviews [43]. While critiques have been raised about their performance due to the limitations associated with the detection and interpretation of ctDNA, as discussed in this review, AI has the potential to overcome these challenges and strengthen CDSS capabilities. Such as the case of the GalleriTM MCED blood test as previously discussed, designed to detect more than 50 different cancer types [40]. Recently concerns have been raised about its sensitivity in detecting cancers. However, a certain commitment in addressing these limitations and advancing towards more sensitive tests became evident in the recent partnership with an AI company, which aims to harness the power of AI to enhance sensitivity and improve the performance of liquid biopsy tests [44].
Recently some AI-based CDSS have been developed also for the management of HCC patients. Choi et al., developed a CDSS algorithm that can recommend the optimal initial treatment of patients with HCC and predict their OS after treatment by the use of AI and machine learning techniques to integrate various patient and tumor-related variables [38]. This study discussed the importance of a good-quality database and selecting pretreatment variables to yield clinically meaningful results. It also highlighted the potential benefits of incorporating genetic information and imaging data to enable more personalized treatment approaches for individual patients [38]. Overall, the study was able to demonstrate the potential of AI and machine learning in improving decision-making in HCC. Another study that highlighted the use of AI in HCC was the study by Oestmann et al., which demonstrated that AI-based image analysis has the potential to expand the role of imaging-based diagnosis in primary liver cancer [45]. In this study, they used a deep learning model, specifically a convolutional neural network (CNN) to differentiate between histopathological validated HCC and non-HCC lesions on multi-phasic contrast-enhanced MRI, including lesions with atypical imaging features. Their model was able to achieve an overall accuracy of 87.3 % [45].
As far as we know, there is no information on the use of AI-based CDSS related to ctDNA or other omic data. This represents the future development for clinical hepatic oncology having the great potential to improve patient outcomes, optimize treatment strategies, and personalize liver cancer management (Fig. 1).
6ConclusionsIn conclusion, ctDNA analysis holds solid potential in HCC care. The use of ctDNA as a non-invasive alternative presents an effective solution to the limitations of tissue biopsy, including invasiveness, tumor heterogeneity, and limited tumor content. The diagnostic potential of ctDNA becomes evident in its capability to detect early-stage HCC through the analysis of methylation patterns and specific gene mutations. Additionally, ctDNA has proven to be a valuable prognostic indicator, correlating with tumor burden, disease progression, and treatment response.
AI holds the transformative potential to revolutionize the field of liquid biopsy for liver cancer. By addressing challenges like low DNA yield and the difficulty in deciphering mutation signals, AI algorithms can enhance sensitivity, improve signal-to-noise ratios, integrate multi-omics data, and develop clinical decision support systems. This convergence of AI and liquid biopsy expedites discoveries and advancements within liver cancer diagnostics and treatment strategies.
Author contributionsInah Marie Aquino reviewed the literature and drafted the manuscript; Devis Pascut reviewed the literature and made critical revisions related to the important intellectual content of the manuscript; Devis Pascut made the final approval of the manuscript submitted.
FundingInah Marie Aquino was supported by a scholarship from the DOST-PCHRD of the Philippines. Devis Pascut was supported by abd intramural grant from Fondazione Italiana Fegato.
The authors would like to express our gratitude to Professor Claudio Tiribelli for his valuable insights and critical reading of the manuscript.



