





Metabolic dysfunction-associated steatotic liver disease (MASLD) is characterized by overweight/obesity, and the presence of type 2 diabetes mellitus is the most important criterion. We propose an independent disease perspective without exclusion criteria and with less heterogeneity and greater impact because, according to the National Health and Nutrition Survey (ENSANUT), in Mexico, 25 % of adults over 60 years of age suffer from diabetes, and 96 % of those over 50 years of age have abdominal obesity. Due to the impact of insulin resistance in the pathophysiology of MASLD, which results in damage to hepatocytes, this work aims to provide an overview of the action pathways of hypoglycemic agents such as glucagon-like-1 receptor agonist and peroxisome proliferator-activated receptor-gamma agonists, whose importance lies in the fact that they are currently undergoing phase 2 studies, as well as dipeptidyl peptidase 4 inhibitors and sodium-glucose co-transporter type 2 inhibitors, which are undergoing phase 1 study trials.
Non-alcoholic fatty liver disease (NAFLD) was renamed metabolic dysfunction-associated steatotic liver disease (MASLD) in an attempt to highlight an independent disease perspective without exclusion criteria because MASLD can coexist with other chronic liver diseases [1]. Furthermore, it is a more specific term and provides an affirmative non-stigmatizing description of the condition rather than a diagnosis of exclusion [2].
MASLD proposes to define metabolic dysfunction as its basis, resulting in a greater disease impact and less heterogeneity, where overweight/obese patients and the presence of type 2 diabetes mellitus (T2DM) will be the most important criteria. According to the National Health and Nutrition Survey (ENSANUT), 25 % of adults over 60 years of age suffer from diabetes, and 96 % of those over 50 years of age have abdominal obesity; therefore, MASLD represents an important issue for Mexico [3].
Furthermore, the dynamic interaction between genetics and environmental factors plays an important role in the prevalence and natural course of MASLD, for which it is worth analyzing the different pathways of metabolic dysfunction that lead to hepatic steatosis [4–6].
For the diagnosis of MASLD, an inclusion of positive criteria was proposed, including evidence of hepatic steatosis, obesity or T2DM, a body mass index less than 25 kg/m2 with two metabolic abnormalities, alterations in blood pressure and abdominal circumference, increased triglyceride levels, decreased HDL cholesterol levels, prediabetes data, and an insulin-resistance index (HOMA-IR) greater than 2.5 or protein C levels greater than 2 mg/dL [1,6]. However, the exclusion of other chronic liver diseases or significant alcohol intake is not required for the diagnosis (Fig 1).

MASLD: new term and shared features with NAFLD.
With MASLD being a relatively new term, there is not enough information about its epidemiology; however, it has been seen that the prevalence of NAFLD in patients with T2DM was 56.8 % [7,8] and T2DM was present in 85 % of the patients diagnosed with NAFLD [9], associated with the fact that most of these patients meet the criteria for the definition of metabolic syndrome, which is the mainstay of the diagnosis of MASLD. The growing epidemic of T2DM and obesity will fuel an increasing prevalence of MASLD worldwide, which shows the importance of investigating pharmacological treatment [4]. This work aims to provide an overview of the main action pathways of several types of hypoglycemic agents that are currently undergoing phase study trials.
2Metabolism pathways of MASLDThe pathophysiology of MASLD is multifactorial and includes inflammatory processes, lipotoxicity, and fibrosis. For that reason, it is important to understand that after food intake, the increase in blood glucose induces the release of insulin from the pancreas, which travels to the liver through the portal circulation, where it begins its activity by storing glucose in the form of glycogen through glycogenesis. Subsequently, it causes the synthesis of fatty acids through the conversion of acetyl Co-A. These anabolic functions induced by insulin activate specific intracellular signals.
On the other hand, the mitochondria play an important role in the regulation of metabolism since they modulate oxidation, ATP synthesis, and the creation of reactive oxygen species [10,11]. The increase in anabolic reactions caused by hepatic insulin resistance leads to an increase in the concentration of free fatty acids, the development of insulin resistance in adipose tissue, and increased lipogenesis [12–14]. This accumulation of fatty acids in the liver is the most implicated event; thus, its activation is carried out through lipogenesis with the participation of transcription factors. Beta-oxidation occurs in the liver, specifically in the mitochondria, where they are transformed into triglycerides and exported to the systemic circulation as constituents of very-low-density lipoproteins.
When either of the two mechanisms is saturated, triglycerides begin to accumulate as lipid droplets in hepatocytes, leading to steatosis [15,16].
Excessive beta-oxidation of free fatty acids results in the production of reactive oxygen species and cytotoxic species. In turn, this excessive production of reactive oxygen species results in oxidative stress that, together with the endoplasmic reticulum, activates the inflammasome, leading to hepatocellular damage, apoptosis and the release of inflammatory mediators [17–19]. According to inflammatory cytokines, transforming growth factor-beta, tumor necrosis factor-alpha (TNFα), interleukin-6 (IL-6), and interleukin-8 have been most frequently implicated in this mechanism since they promote the movement of polymorphonuclear and mononuclear leukocytes towards the inflamed tissue, thus activating liver stellate cells and generating increased collagen synthesis and fibrosis development [20–23].
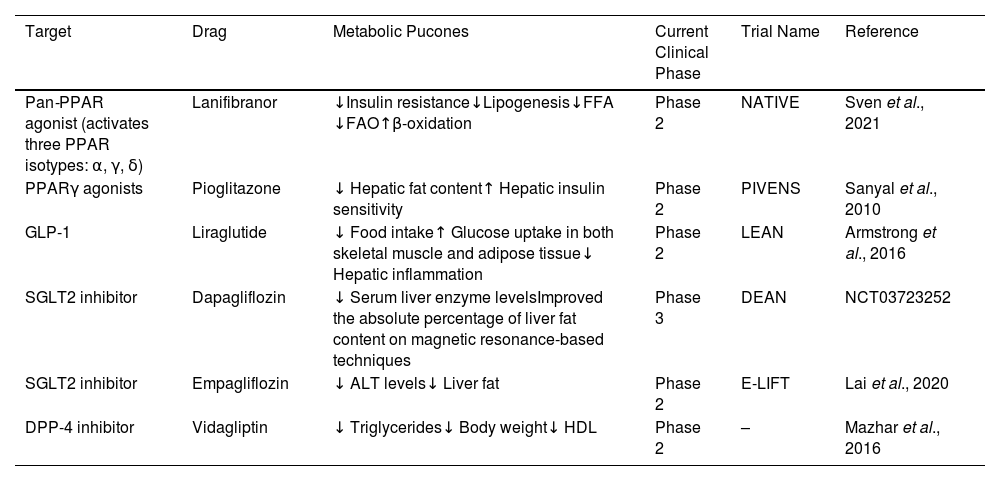
3Relationship between hypoglycemic agents and MASLDDue to the relationship of MASLD with metabolic disturbances [9], both conditions share pathophysiological characteristics, such as insulin resistance, which results in tissue damage to hepatocytes through different ways in which the efficacy of hypoglycemic agents has been evaluated to reduce liver fat and improve liver injury [24]. The hypoglycemic agents that have been studied to achieve the most favorable results are glucagon-like-1 receptor agonists (GLP-1RAs) and peroxisome proliferator-activated receptor-gamma agonists (PPARγ), which are currently undergoing phase 2 studies. This phase 2b placebo-controlled randomized clinical trial is the NATIVE trial, where lanifiranor, a first-in-class pan-PPAR agonist, was tested in patients with non-alcoholic steatohepatitis (NASH). The study demonstrated that a 1200 mg dose of lanifibranor decreased the histologic steatosis, activity, and fibrosis (SAF) score by at least two points, so this drug seems to be the most promising treatment option. However, the findings will be confirmed in phase 3 clinical trials [25] (Table 1).
Summary of controlled trials and outcomes of glucose-lowering drugs.
| Target | Drag | Metabolic Pucones | Current Clinical Phase | Trial Name | Reference |
|---|---|---|---|---|---|
| Pan-PPAR agonist (activates three PPAR isotypes: α, γ, δ) | Lanifibranor | ↓Insulin resistance↓Lipogenesis↓FFA ↓FAO↑β-oxidation | Phase 2 | NATIVE | Sven et al., 2021 |
| PPARγ agonists | Pioglitazone | ↓ Hepatic fat content↑ Hepatic insulin sensitivity | Phase 2 | PIVENS | Sanyal et al., 2010 |
| GLP-1 | Liraglutide | ↓ Food intake↑ Glucose uptake in both skeletal muscle and adipose tissue↓ Hepatic inflammation | Phase 2 | LEAN | Armstrong et al., 2016 |
| SGLT2 inhibitor | Dapagliflozin | ↓ Serum liver enzyme levelsImproved the absolute percentage of liver fat content on magnetic resonance-based techniques | Phase 3 | DEAN | NCT03723252 |
| SGLT2 inhibitor | Empagliflozin | ↓ ALT levels↓ Liver fat | Phase 2 | E-LIFT | Lai et al., 2020 |
| DPP-4 inhibitor | Vidagliptin | ↓ Triglycerides↓ Body weight↓ HDL | Phase 2 | – | Mazhar et al., 2016 |
PPAR: peroxisome proliferator-activated receptor; FFA: free fatty acid; FAO: fatty acid oxidation; GLP-1: glucagon-like peptide-1; SGLT2: sodium-glucose cotransporter 2; DPP-4: dipeptidyl peptidase 4 inhibitor; HDL: high-density lipoprotein; ALT: alanine transaminase.
The most recommended treatment in MASLD patients with obesity or with T2DM is with GLP-1RA molecules, where, in a phase 2 randomized controlled trial, it was demonstrated that there was a significant improvement in the absolute percentage of liver fat content through magnetic resonance-based techniques using different GLP-1 RAs, especially liraglutide and semaglutide [26]. In the LEAN trial, a small phase 2b trial, treatment with 1.8 mg/day liraglutide resulted in a histologic resolution of NASH and a decreased progression of fibrosis [27]. Meanwhile, dipeptidyl peptidase 4 (DPP-4) inhibitors and sodium-glucose co-transporter type 2 (SGLT2) inhibitors are undergoing phase 2 study trials [28] (Table 1).
The European Association for the Study of the Liver (EASL) suggests that drug treatment should be considered for patients with NASH (stage F2 or higher) or with a high risk for disease progression, such as patients with T2DM or metabolic syndrome. Pioglitazone improves liver histology in patients with and without DM2 with biopsy-proven NASH. Although other drugs have not been withdrawn from phase 3 trials yet, the EASL suggests that using pioglitazone in these patients may improve histologic features in steatosis and possibly in fibrosis [29]. These findings were proved in the PIVENS trial, which compared low-dose pioglitazone versus a placebo for 2 years in patients without overt diabetes [30], as well as in another placebo-controlled trial including subjects with NASH for whom diet and pioglitazone improved glycemic control and glucose tolerance, normalized liver aminotransferase levels, decreased hepatic fat content, and increased hepatic insulin sensitivity [31] (Table 1). However, the American Association for the Study of Liver Diseases (AALSD) recommends that pharmacological treatments should generally be limited to those with biopsy-proven NASH and fibrosis. Then, the importance of a weight loss between 7 and 10 % is needed to improve most of the histopathological features of NASH, including fibrosis. [32].
Similarly, the American Association of Clinical Endocrinology (AACE) mentions that in addition to pioglitazone, GLP-1RAs are recommended for patients with T2DM and biopsy-proven NASH. These drugs offer additional cardiometabolic benefits in patients with T2DM and NAFLD. Finally, due to the lack of evidence of efficacy, metformin, acarbose, dipeptidyl peptidase IV inhibitors, and insulin are not recommended for the treatment of NAFLD [33]. Instead, biguanides, especially metformin, have become the preferred first-line oral blood-glucose-lowering agent to manage T2DM, as they reduce hepatic glucose production by decreasing hepatic gluconeogenesis [34].
Metformin's mechanism of action is based on the interruption of mitochondrial oxidative processes, resulting in a reduced AMP/ATP ratio and, subsequently, in the activation of activated protein kinase (AMP), a major cell regulator of lipid and glucose metabolism [35]. The activation of AMP in the liver stimulates the β-oxidation of fatty acids and inhibits de novo synthesis, thus potentially leading to reduced liver steatosis [36]. Also, to protect against lipid accumulation at the cellular level, metformin has an effect on the nuclear exclusion of Forkhead box protein O1, reducing the expression of fatty-acid-binding protein 4 [37]. The effect of metformin on liver histology remains unclear, but it has been shown that it contributes to reducing ALT and AST levels; however, it seems that it does not have a meaningful impact on liver histology [38].
4PPARγ agonistsAmong the receptors and targets of hypoglycemic drugs that influence pharmacodynamics, PPARγ is one of the receptors for nuclear hormones that regulates the transcription of genes involved in cell growth, differentiation, and metabolism in response to lipophilic hormones, dietary fatty acids, and their metabolites [39–41]. Genes activated by PPARγ stimulate the lipid uptake and adipogenesis of fat cells [42]. Within this group of drugs, thiazolidinediones such as pioglitazone significantly improve liver steatosis and lobular inflammation independently of the presence of T2DM. In fact, thiazolidinediones increase the storage of fatty acids in adipocytes, causing excess fat in the liver to be deposited in the adipose tissue.
PPAR-γ is one of the three isotypes that have been identified in vertebrates. The gene is encoded by six exons distributed in different domains with different functions; within these domains, the most significant is the ligand-binding domain, which facilitates adherence to a specific DNA consensus region called the peroxisome proliferator response element. This binding is carried out after the activation of PPAR-γ and its subsequent heterodimer formation with the retinoid X receptor [43,44].
Moreover, PPARγ has two isoforms, 1 and 2, to which it can be spliced, most of which are found in adipose tissue, the colon, and macrophages [45]. The PPARy2 isoform has been shown to play an important role in adipogenesis and in regulating the expression of adipose genes such as adipocyte protein 2 and phosphoenolpyruvate carboxykinase [46]. In addition, the pharmacodynamics of PPARγ agonists are extensively metabolized by cytochrome (CYP) P450 enzymes. Specifically, pioglitazone is mainly metabolized by CYP2C8, while CYP3A4 / 5 and CYP1A1 contribute to its metabolism to a lesser extent [47,48].
5GLP-1 RAsGastric inhibitory polypeptide (GIP) and glucagon-like-1 (GLP-1) are types of incretin receptors that are secreted after food intake and integrate nutrient-derived signals to control the amount of food eaten and energy absorption. Their secretion is carried out from the L cells, which are found in the colon and distal ileum [49]. On the other hand, gastric inhibitory peptide is secreted through K cells, which are found mainly in the jejunum and duodenum. Its mechanism of action relies on binding to its receptor in pancreatic beta cells, generating an increase in the levels of cyclic adenosine monophosphate, which results in glucose-dependent insulin secretion and proliferation [41,45,50].
Both GLP-1 and GIP represent up to 70 % of insulin secretory responses after nutrient ingestion [51]. These two polypeptides carry out their action when they are coupled to G protein-coupled receptors. GIP associates with its receptor, which is found mainly in the beta cells of the islets in addition to being in the central nervous system. On the other hand, GLP-1RAs are found in both α cells and β cells in peripheral tissues such as the heart, lungs, kidneys and gastrointestinal tract [52).
It has been seen that in the pathophysiology of MASLD, GLP-1 decreases the response to oxidative stress in the endoplasmic reticulum and improves steatosis data by acting directly on hepatocytes, modulating lipid metabolism and liver insulin signaling [53,54]. Similarly, in individuals with T2DM, GLP-1 analogs reduce the frequency of inflammatory macrophages and decrease the levels of the inflammatory cytokines TNFα, IL-6 and interleukin 1-β; this effect is independent of changes in glycemic and body weight control [12,22].
Among the molecular signals that explain the role of GLP-1 in hepatocytes, the protein kinase B (Akt) signaling pathway has been seen involved in steatotic hepatocytes, since the phosphorylation of AkT is decreased. This protein is responsible for the regulation of lipid and glucose metabolism; thus, an increase in the phosphorylation of AkT improves the up-regulation of key elements of the insulin signaling pathway of hepatocytes [55,56]. A decrease in the anti-apoptotic protein B-cell lymphoma 2 (Bcl2) was also found, which resulted in an increase in the Bcl2-like protein 4 (BAX) (proapoptotic) / Bcl2 ratio in the liver tissue of patients with MASLD [57].
With these data, it has been suggested that insulin resistance in the hepatocyte may be the cause of cell apoptosis [56]. For example, in the sub-analysis of the AWARD program, it was shown that dulaglutide, an incretin analog, significantly reduces the serum activity of transaminases and decreases the levels of gamma-glutamyl transpeptidase compared with a placebo. This study was carried out in patients with T2DM to study the decrease in liver fat [58].
Recent studies in animals confirmed that GLP-1RAs, weight loss and hypoglycemic effects can reduce inflammatory lesions of the liver and even delay the process of change from steatosis to fibrosis [59]. Furthermore, in vitro studies of Langerhans islets from neonatal rats have reported a greater inhibition of apoptosis induced by free fatty acids with Liraglutide, a GLP-1 receptor agonist, thus increasing the mass of beta cells and their differentiation [60].
6GLT2 inhibitorsAnother group of drugs that has been studied is SGLT-2, which act by inhibiting the reabsorption of glucose in the proximal tubule of the nephron, causing glycosuria and a reduction in glucose levels in the plasma [61]. The changes seen in SGLT-2 inhibitors can be explained by various mechanisms such as the suppression of adipocyte lipolysis, which aids in the inhibition of the accumulation of ectopic fat in the liver. Another mechanism involves beta-oxidation in the liver and the secretion of very-low-density lipoproteins into the circulation through the up-regulation of carnitine palmitoyltransferase I, a mitochondrial enzyme that regulates the membrane transport of free fatty acids, PPARα and microsomal triglyceride transfer protein genes in hepatocytes, which causes a progression in inflammation and liver fat accumulation [62,63].
These drugs have become therapeutically promising in NAFLD, as several pilot studies suggested a reduced transaminase activity, body weight, fatty liver index and liver histology according to steatosis and fibrosis. Two open-label, randomized trials conducted in Japan compared the efficacy of SGLT-2 inhibitors with other oral anti-glycemic agents such as pioglitazone and metformin, among others. Within the results, it was found that the liver fat content decreased significantly in the group that received luseogliflozin, an SGLT2 inhibitor, compared with that in the metformin group [64]. In another study comparing the efficacy of ipragliflozin against pioglitazone in diabetic patients with NAFLD, there was a significant reduction in serum ALT levels, glycated hemoglobin, and plasma glucose in the two groups, where the group treated with ipragliflozin had a significant reduction in visceral fat and weight loss [65,66].
Moreover, the E-LIFT study investigated the response of empagliflozin, another SGLT2 inhibitor, in patients with T2DM with NAFLD. The amount of fat in the liver was evaluated after 20 weeks of treatment. The fat in the livers of patients with empagliflozin decreased significantly from 16.2 % to 11.3 %; in contrast, the control group decreased in a range from 16.4 % to 15.6 % [14]. SGLT2 inhibitors such as empagliflozin reduce liver fat, improving ALT levels in patients with T2DM and NAFLD. Empagliflozin was implicated in a pilot study concluding that it reduces liver fibrosis in patients with T2DM [67] (Table 1).
Various studies have been carried out comparing the results of each of these groups. Interestingly, in a meta-analysis, 26 studies were included in which the drugs that demonstrated a change in hepatic steatosis were GLP-1 agonists and pioglitazone. They concluded that among the four antidiabetic drugs, pioglitazone, SGLT2 inhibitors, GLP-1 agonists and −4 inhibitors, pioglitazone produces significant improvements in liver histology and enzyme levels in NAFLD patients [68].
7Dipeptidyl peptidase-4 (DPP-4) inhibitorsDPP-4 inhibitors act primarily by blocking the enzyme DPP-4, which is an aminopeptidase that is widely expressed in many tissues, such as the liver, lungs, kidneys, intestinal brush border membranes, lymphocytes, and endothelial cells [55]. The purpose of this group of hypoglycemic agents is to increase serum levels of incretin hormones such as GLP-157 and gastric inhibitory polypeptide, which is accomplished by inhibiting the cell surface enzyme serine aminopeptidase DPP-4, as it normally degrades and rapidly inactivates GLP-1, GIP, and other in vivo peptides by cleaving the two n-terminal amino acids [69]. The inactivation of incretins through DPP-4 activity results in only 10–20 % of the total biologically active plasma GLP-1 [52].
Moreover, increased levels of DPP-4 have been reported in patients with hepatic steatosis and also with apoptosis of the hepatocytes, for which it has been proposed that DPP-4 inhibitors improve the histopathological findings of hepatic steatosis [53]. Among the concerns about these medications, they found their association with an increased risk of suffering cholangiocarcinoma (hazard ratio (HR) 1.77, 95 % CI 1.04–3.01) [70].
8Discussion and conclusionsThe concept of MASLD possesses less heterogeneity and the advantage of being capable of coexisting with other diseases. Nowadays, several successful studies have focused on pharmacological treatments related to hepatic steatosis in metabolic dysfunction and their approach through different pathways, either by modulating fatty acid storage and glucose metabolism, such as with PPARγ-gamma or GLP-1 agonists, or using DPP-4 inhibitors, which have a protective effect on hepatocytes by modulating lipid metabolism and hepatic insulin signaling, then minimizing the response to oxidative stress in the endoplasmic reticulum (Fig. 2).

Pathways of action of hypoglycemic agents in the liver and their relationship with the pathophysiology of MASLD.
According to the current scenario, alterations in pathways such as phosphatidylinositol-3 kinase (PI3-k)/Akt contribute to liver damage by generating apoptosis in the hepatocytes and releasing inflammatory cytokines. Moreover, the excessive beta-oxidation of free fatty acids due to lipogenesis with the involvement of transcription factors such as PPAR-γ agonists achieves the regulation of adipose gene expression. Several pathways have been investigated in patients with diagnostic criteria for MASLD regarding their effect at the molecular level in hepatocytes. However, more studies are still needed to compare the different therapies with different groups of patients according to the diagnostic criteria of MASLD, and consider several criteria such as gender, age, and genetic and epigenetic factors, as well as studies that involve research on insulin signaling pathways.
Author contributionsACMR: design concept, acquisition of information and writing the manuscript; JMZV: critical revision and correction of the manuscript for important intellectual content; MU: critical revision of the manuscript; VJBB: guidance for design, interpretation of information and correction of the manuscript.
FundingThis research did not receive any specific grants from funding agencies in the public, commercial or not-for-profit sectors.
This article has no contributions to acknowledge.



