



Interferon-α (IFN) has shown potential benefits in patients with hepatocellular carcinoma (HCC), and these effects may be mediated by inhibiting cancer cell proliferation. However, the detailed mechanisms underlying the anti-proliferative effects of IFN remain obscure. In this study, we evaluate the role of the novel oncogenic microRNA (miRNA) miR-155 in the anti-proliferative effects of pegylated interferon-α (PEG-IFN) on HCC cells.
MethodsThe effects of PEG-IFN on HepG2 cell proliferation, migration and invasion were determined using the MTT assay, flow cytometry analysis and the Transwell assay, respectively. Reverse transcription quantitative polymerase chain reaction was used to analyze miR-155 expression. The levels of proteins involved in Wnt/β-catenin signal transduction were determined by western blot analysis and immunofluorescence staining. Mimics of miR-155 were transfected into HepG2 cells to assess the role of miR-155 in the PEG-IFN-induced anti-proliferative effect.
ResultsPEG-IFN significantly inhibited the proliferation, migration and invasion of HepG2 cells in a dose-dependent manner by inhibiting cell cycle progression. In parallel with reduced cell proliferation, migration and invasion, miR-155 was efficiently downregulated by PEG-IFN in a dose-dependent manner. Moreover, the transfection of miR-155 decreased the inhibitory effect of PEG-IFN on HepG2cell proliferation, migration and invasion, as well as the downregulation of proteins in the Wnt/β-catenin pathway.
ConclusionsThe anti-proliferative effects of PEG-IFN on HCC are at least partially attributable to the downregulation of miR-155.
Hepatocellular carcinoma (HCC) is the dominant pathological type of primary liver cancer and the third leading cause of cancer-related death worldwide [1]. Despite recent advances in the diagnosis and treatment of HCC, including hepatectomy and liver transplantation, the prognosis of patients with HCC remains poor due to the high rates of recurrence and early metastasis [2]. Studies have reported various effects of interferon-α (IFN)therapy on HCC in patients with chronic viral hepatitis, ranging from effectively preventing the development of HCC to reducing recurrence after initial curative therapy by liver resection or radiofrequency ablation [3]. Furthermore, interferon therapy significantly reduces the risk for the development of HCC, regardless of whether a sustained virological response occurs [4,5]. Thus, IFN may exert other antitumour effects in addition to decreasing the viral load in these patients, potentially by inhibiting cancer cell proliferation [6,7]. Although its molecular mechanisms of action are not yet completely understood, the anti-proliferative activity of IFN makes it an intriguing option for the chemoprevention and chemotherapy of HCC.
MicroRNAs (miRs), evolutionarily conserved gene expression regulators, participate in many fundamental physiological processes. In cancer development, miR functions as a tumour suppressor or an oncogene by targeting specific genes in a 3′UTR-dependent manner. The overexpression of oncogenic miRNAs (oncomiRs) downregulates the expression of tumour suppressors and/or other genes involved in cell differentiation, leading to tumour formation by stimulating proliferation, angiogenesis and invasion [8]. Among the known oncomiRs, miR-155 has been reported to be involved in the development of a number of types of malignancies, such as B cell malignancies, breast cancer, colon cancer and HCC [9]. Notably, miR-155 is an oncomiR that is frequently upregulated during the pathogenesis of HBV, HCV and non-alcoholic steatohepatitis (NASH)-associated HCC [10]. According to other studies, miR-155 is involved in the initiation of hepatocarcinogenesis and is associated with poor clinicopathological features and reduced survival of patients with HCC [11,12]. Moreover, dysregulation of the Wnt/β-catenin signalling pathway is observed in HCC. Accumulating evidence has indicated the importance of the regulatory network of miR-155 and the Wnt/β-catenin pathway in HCC development [13]. The upregulation of miR-155 expression in patients with hepatitis and HCC has been shown to promote hepatocyte proliferation and HCC tumour growth by activating the Wnt/β-catenin signalling pathway [14]. Based on these data, miR-155 represents a promising target for the diagnosis and treatment of HCC. Therefore, new targeted therapeutic strategies for HCC based on the regulatory network of miR-155 and the Wnt/β-catenin pathway are urgently needed. However, the use of anti-miRs as therapeutic agents for miR-155-overexpressing cancers has been hindered by the instability of anti-miRs and by targeting issues with nanodelivery systems [15–17]. Pegylated interferon-α (PEG-IFN) represents an attractive and stable alternative. The expression of miR-155 is significantly decreased in patients with hepatitis C who are treated with IFN and ribavirin, and the lowest miR-155 expression was detected in patients who lacked HCV RNA in both serum and peripheral blood mononuclear cells [18]. As shown in our previous study, PEG-IFN inhibits hepatic progenitor cell proliferation and hepatocarcinogenesis by modulating the Wnt/β-catenin pathway [19]. However, researchers have not clearly determined whether miR-55 is involved in the anti-proliferative effects of PEG-IFN.
Based on these findings, we aimed to investigate the molecular mechanism underlying the anti-proliferative effects of PEG-IFN on HepG2 cells in vitro. PEG-IFN inhibits HepG2 cell proliferation, and this anti-proliferative effect is at least partially mediated by the downregulation of miR-155, which may suggest a novel method for treating HCC. A better understanding of its role in HCC cells may lead to the successful manipulation of liver biology for therapeutic purposes.
2Materials and methods2.1Cell lines and cultureThe human HCC cell line HepG2 was obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM, which was adjusted to contain 4mM l-glutamine, 1.5g/L sodium bicarbonate, 4.5g/L glucose, 10% FBS, 100units/mL penicillin, and 65units/mL streptomycin. All cell lines were maintained at 37°C in a humidified incubator containing 5% CO2. Cultured cells were treated with PEG-IFN in complete DMEM.
2.2Drug treatmentsPEG-IFN (Pegasys®; Roche Pharmaceuticals Corp., Shanghai, China) was added to cells at increasing concentrations (3.6–3600ng/mL) for 48h in serum-free medium.
2.3MTT assayCell viability was determined using the MTT assay. Briefly, the cells were seeded in 96-well dishes at a density of 1×104 cells per well and treated with different concentrations of PEG-IFN. Then, each well was supplemented with 10μL of MTT (Sigma–Aldrich) and incubated at 37C for 4h. The medium was then removed, and 150μL of DMSO (Sigma–Aldrich, Shanghai, China) was added to solubilize the MTT formazan crystals. The optical density was measured at 490nm.
2.4Cell transfectionThe miR-155 mimics and negative controls (NC) were designed and synthesized by GenePharma (Shanghai, China). HepG2 cells were seeded into each well of a 6-well plate, incubated overnight and then transfected with the miR-155 mimic or negative control using Lipofectamine 2000 (Invitrogen, Shanghai, China) according to the manufacturer's instructions to selectively upregulate miR-155. Cells were harvested 24h after transfection for further analysis.
2.5Quantitative RT-PCR analysis of miR-155 expressionAfter treatment with or without PEG-IFN for 48h, HepG2 cells were collected, and miRNAs were extracted with Trizol reagent (Qiagen, Germany) using a standard method. PrimeScript™ RT Master mix was used to reverse transcribe RNA samples into cDNAs according to the manufacturer's protocol. qRT-PCR was performed using the miScript SYBR Green PCR Kit (Qiagen, Germany) and an ABI PRISM 7700 cycler (Applied Biosystems, USA). The amplification reactions were as follows: 95°C for 15min, followed by 40 cycles of 94°C for 15s, 55°C for 30s and 70°C for 30s. The primers used in this study were as follows: miR-155, forward 5′-CGGTTTAATGCTAATCGTGA-3′ and reverse 5′-GAGCAGGGTCCGAGGT-3′; U6, forward 5′-CTCGCTTCGGCAGCACA-3′ and reverse 5′-AACGCTTCACGAATTTGCGT-3′. The relative level of miR-155 was calculated and subsequently converted to a fold change using the 2–ΔΔCT method.
2.6Transwell migration assay and Matrigel invasion assayFor Transwell migration assays, the cells were counted, and 2×104 cells in 500μL of serum-free DMEM containing the indicated concentration of PEG-IFN were seeded into the upper part of each chamber, whereas the lower compartments were filled with DMEM supplemented with 10% foetal bovine serum as a chemo-attractant. For invasion assays, cells were plated in the top chamber on a Matrigel-coated (100μg/cm2) Transwell membrane (8-μm pore size, BD Biosciences, USA), and the remaining procedures were similar to those of the Transwell migration assays. Following an 18h incubation at 37°C, cells on the upper surface of the filter that had not invaded the Matrigel were removed with a cotton swab. The invaded cells on the lower surface of the filter were fixed with formaldehyde (4%) and stained with 0.1% crystal violet in 2% methanol. Invasion was determined by counting the cells in five microscopic fields from each well, and the extent of invasion was expressed as an average number of cells per microscopic field.
2.7Western blotsAfter treatment with or without PEG-IFN for 48h, HepG2 cells were collected. Proteins were extracted in lysis buffer (50mM Na2HPO4, 50mM NaH2PO4, 0.2M NaCl, 5mM EDTA, and 1% Triton X-100, pH 6.0). After a 30-minincubation on ice, the samples were centrifuged (13,000×g, 20min, 4°C), and 2-dithiothreitol (DTT) loading buffer (0.4M Tris, pH 6.8; 4% SDS; 20% glycerol; and 10% DTT) was added to the sample supernatants and incubated for 5min at 95°C. Following electrophoretic separation by SDS-polyacrylamide gel electrophoresis, proteins were electroblotted onto nitrocellulose membranes. The membranes were blocked with NET buffer (150mM NaCl; 5mM EDTA, pH 8.0; 50mM Tris/HCl, pH 7.5; and 0.05% Triton X-100) containing 2.5% gelatine (Merck) for 1 hat room temperature and then incubated with polyclonal antibodies against β-catenin (Santa Cruz, CA, USA), cyclin D1 (Santa Cruz, CA, USA), C-myc (Santa Cruz, CA, USA), and adenomatous polyposis coli (APC) (Santa Cruz, CA, USA) for 1 hat room temperature. Thereafter, the membranes were washed with NET buffer and then incubated with a peroxidase-conjugated antibody at a dilution of 1:20,000. Antibody binding was visualized using a Hydrogen Peroxidase-Chemiluminescence Detection Kit (Frontier Laboratories, Koriya-ma, Japan). A semiquantitative evaluation of the bands was performed by densitometry. The levels of β-catenin protein were normalized to the levels of the housekeeping protein β-actin.
2.8Flow cytometry analysisThe cell cycle was analyzed as previously described. Briefly, aliquots of cells (1×106) were pelleted (1300rpm for 5min at 4°C) and washed twice with ice-cold phosphate-buffered saline (PBS). The cells were then fixed with 70% ethanol overnight at 4°C, washed with PBS and then digested with DNase-free RNase A (10μg/mL) at 37°C for 30min. Prior to FACS analysis, the cells were resuspended in 200μL of propidium iodide (PI, 10μg/mL; Sigma, St Louis, MO, United States) to label the DNA. A BD FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, United States) flow cytometer was used to analyze the cellular DNA contents.
2.9Immunofluorescence stainingHepG2 cells cultured in the presence or absence of PEG-IFN (3600ng/mL) were fixed with 4% paraformaldehyde at 37°C for 30min. Permeabilization of the cells was achieved after incubation with PBS containing 0.2% Triton X-100 for 30min at 37°C. The cells were blocked with a buffer containing 1% bovine serum albumin for 1h to minimize nonspecific binding of the antibody. The β-catenin antibody (Santa Cruz, CA, USA) was applied at a 1:25 dilution for 90min at 37°C. As a negative control, PBS was used instead of the primary antibody to exclude nonspecific binding of the secondary antibody. No fluorescent labelling was observed in the negative control. After repeated washes with PBS, cells were incubated with a goat-anti-mouse antibody labelled with fluorescein isothiocyanate (1:10) for an additional 30min. Finally, the cell nuclei were counterstained with DAPI. Images were obtained using a confocal laser scanning microscope.
2.10Statistical analysisAll results are reported as the means±SD. Measurement data were analyzed using one-way analysis of variances (ANOVA, SPSS 11.5). P<0.05 was considered statistically significant.
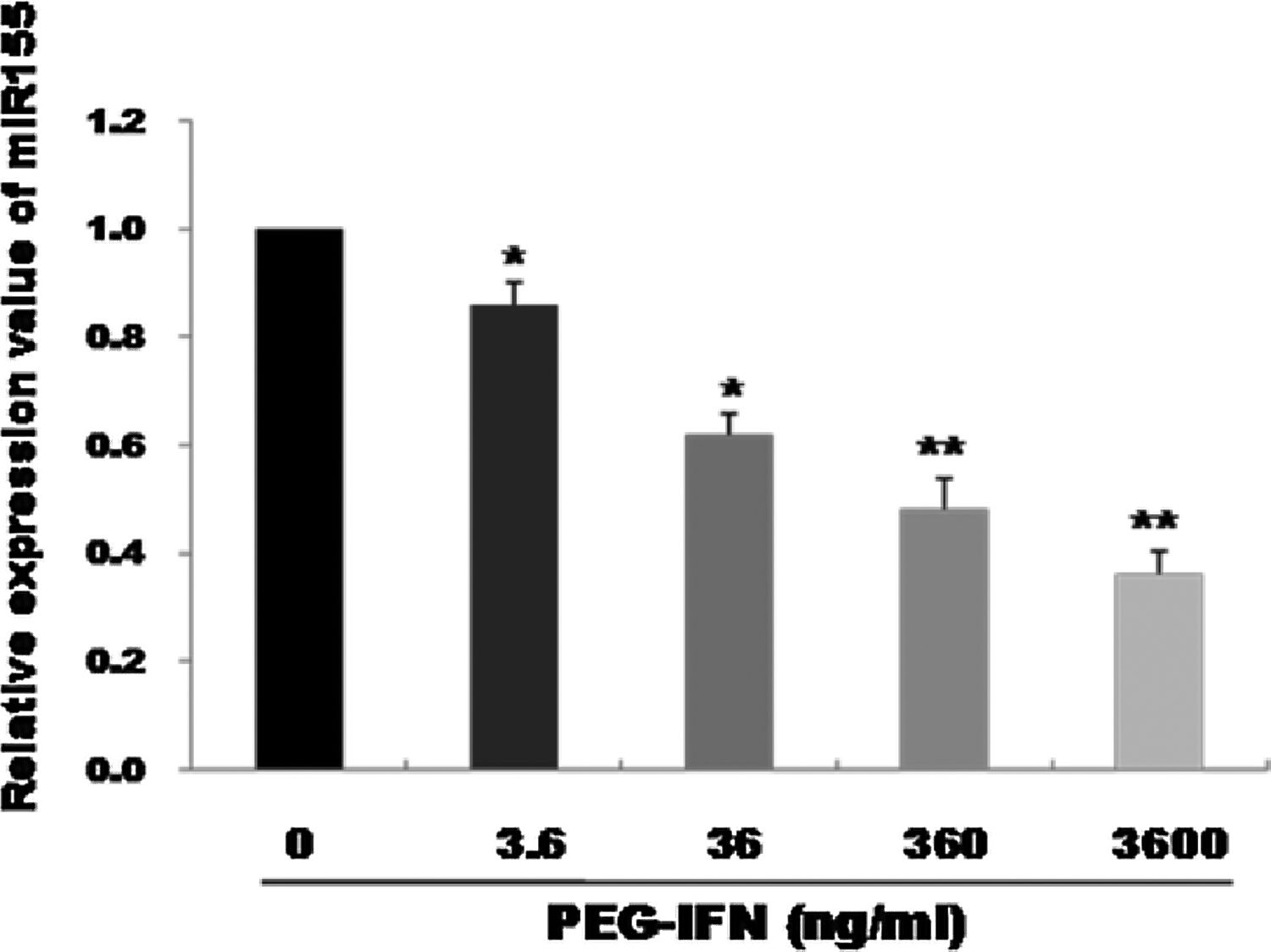
3Results3.1PEG-IFN treatment downregulates miR-155 expression in HepG2 cellsqRT-PCR was used to observe the effects of treatment with different PEG-IFN concentrations on miR-155 expression. As shown in Fig. 1, compared with the control, miR-155 was downregulated in a dose-dependent manner after 48h of treatment with 3.6–3600ng/mL PEG-IFN, and the lowest expression was observed in cells treated with 3600ng/mL PEG-IFN. Thus, PEG-IFN treatment downregulated miR-155 expression.
 of PEG-IFN for 48h. The data represent the means±SD derived from three independent experiments. (*P<0.05, **P<0.01, vs. HepG2 cells not given PEG-IFN).")
miR-155 was down-regulated by PEG-IFN treatment. HepG2 cells were treated with various concentrations (3.6, 36, 360, and 3600ng/ml) of PEG-IFN for 48h. The data represent the means±SD derived from three independent experiments. (*P<0.05, **P<0.01, vs. HepG2 cells not given PEG-IFN).
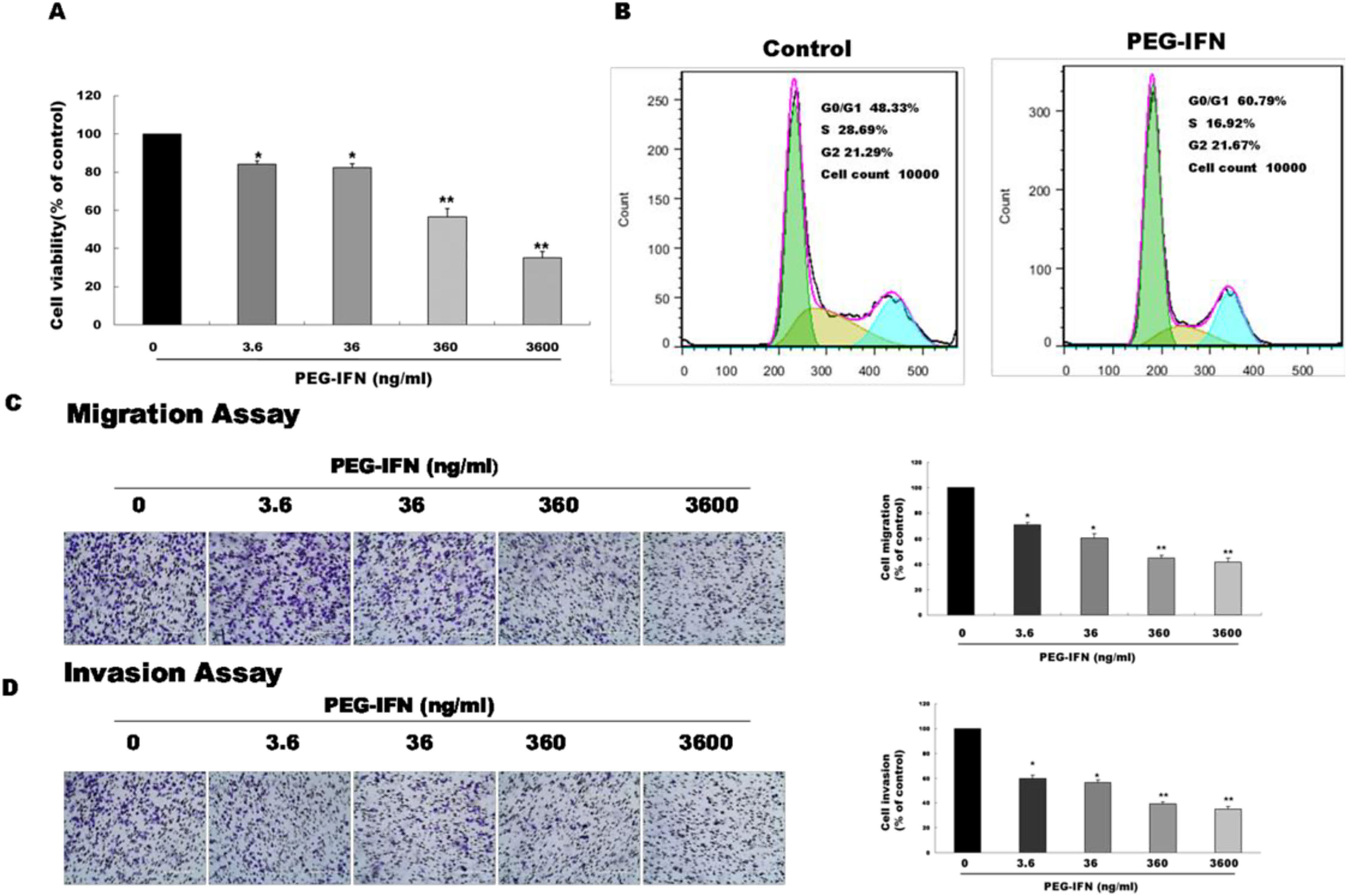
We examined whether PEG-IFN suppressed cell growth by using the MTT assay to investigate the effects of PEG-IFN on HepG2 cells. HepG2 cells were treated with different concentrations PEG-IFN (3.6–3600ng/mL) for 48h. As shown in Fig. 2A, the PEG-IFN treatment markedly inhibited HepG2 cell proliferation. We next performed a cell cycle analysis using flow cytometry in HepG2 cells that had been treated with PEG-IFN (3600ng/mL) for 48h. PEG-IFN increased the percentage of cells in the G1 subpopulation to 60.79% within 48h, compared to 48.3% of unexposed cells (Fig. 2B). In addition, PEG-IFN decreased the percentage of cells in the S phase subpopulation compared with unexposed cells. Moreover, we tested the effect of PEG-IFN on HepG2 cell migration and invasion by performing Transwell migration and invasion assays. Treatment with various concentrations of PEG-IFN for 48h significantly inhibited the migration of HepG2 cells (Fig. 2C). Similarly, PEG-IFN treatment strongly inhibited the invasion of HepG2 cells (Fig. 2D). Based on these results, PEG-IFN inhibited HepG2 cell growth.
 HepG2 cells were treated with various concentration of PEG-IFN for 48h, and the cell viability was measured using MTT assay (*P<0.05, **P<0.01, vs. HepG2 cells not given PEG-IFN). (B) Flow cytometry analysis showed that PEG-IFN induced G1-arrest of HepG2 cells. (C) Exposure to various concentrations of PEG-IFN resulted in dose-dependent migration inhibition of HepG2 cells (*P<0.05, **P<0.01, vs. HepG2 cells not given PEG-IFN). (D) Exposure to various concentrations of PEG-IFN resulted in dose-dependent migration inhibition of HepG2 cells (*P<0.05, **P<0.01, vs. HepG-2 cells not given PEG-IFN).")
PEG-IFN-mediated inhibition of HepG2 cells proliferation, migration and invasion. (A) HepG2 cells were treated with various concentration of PEG-IFN for 48h, and the cell viability was measured using MTT assay (*P<0.05, **P<0.01, vs. HepG2 cells not given PEG-IFN). (B) Flow cytometry analysis showed that PEG-IFN induced G1-arrest of HepG2 cells. (C) Exposure to various concentrations of PEG-IFN resulted in dose-dependent migration inhibition of HepG2 cells (*P<0.05, **P<0.01, vs. HepG2 cells not given PEG-IFN). (D) Exposure to various concentrations of PEG-IFN resulted in dose-dependent migration inhibition of HepG2 cells (*P<0.05, **P<0.01, vs. HepG-2 cells not given PEG-IFN).
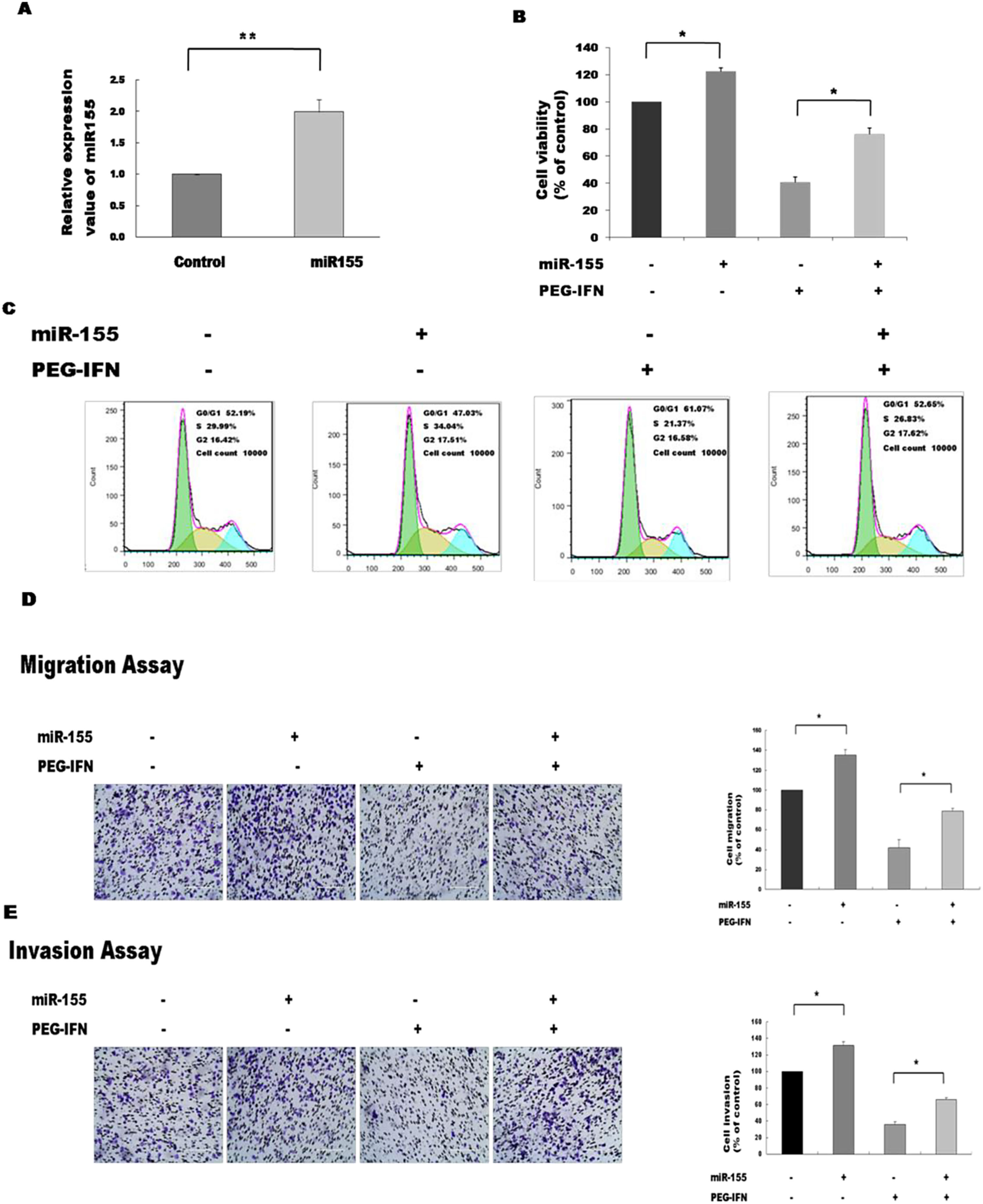
Since PEG-IFN downregulated miR-155 expression and this small RNA is reported to affect the viability of cancer cells, we were interested in determining whether alterations in the expression of miR-155 affected the PEG-IFN-induced inhibition of HepG2 cell growth. To address this question, we overexpressed miR-155 and observed the effects on the activity of PEG-IFN. The expression of miR-155 was significantly elevated in cells transfected with miR-155 mimics, suggesting that miR-155 mimics successfully penetrated the HepG2 cells (Fig. 3A). The transfection of miR-155 mimics reduced the PEG-IFN-mediated inhibition of HepG2 cell proliferation, as evidenced by a significant increase in cell proliferation and a decrease in the population of cells in G1 phase compared to the controls (Fig. 3B and C).
 Transfection of miR-155 mimics significantly increased the expression of miR-155 in HepG2 cells. **P<0.01 indicate significant differences from the control groups B After transfected with miR-155 mimics, the cells were treated with PEG-IFN (3600ng/ml). miR-155 significantly promoted proliferation in HepG2 cells with or without PEG-IFN treatment. *P<0.05 indicate significant differences from the respective control groups. (C) Flow cytometry analysis showed that miR-155 mimics decreased G0/G1 phase cell population with or without PEG-IFN treatment. (D) miR-155 mimics significantly increased migration ability in HepG2 cells with or without PEG-IFN treatment. *P<0.05 indicate significant differences from the respective control groups. (E) miR-155 mimics significantly increased invasion ability in HepG2 cells with or without PEG-IFN treatment. *P<0.05, indicate significant differences from the respective control groups.")
Upregulation of miR-155 abrogates the inhibitory effects of PEG-IFN on HepG2 cells growth. (A) Transfection of miR-155 mimics significantly increased the expression of miR-155 in HepG2 cells. **P<0.01 indicate significant differences from the control groups B After transfected with miR-155 mimics, the cells were treated with PEG-IFN (3600ng/ml). miR-155 significantly promoted proliferation in HepG2 cells with or without PEG-IFN treatment. *P<0.05 indicate significant differences from the respective control groups. (C) Flow cytometry analysis showed that miR-155 mimics decreased G0/G1 phase cell population with or without PEG-IFN treatment. (D) miR-155 mimics significantly increased migration ability in HepG2 cells with or without PEG-IFN treatment. *P<0.05 indicate significant differences from the respective control groups. (E) miR-155 mimics significantly increased invasion ability in HepG2 cells with or without PEG-IFN treatment. *P<0.05, indicate significant differences from the respective control groups.
We investigated whether the inhibitory effects of PEG-IFN on HepG2 cell migration and invasion were associated with miR-155 expression and found that mimic-induced miR-155 overexpression reversed the PEG-IFN-induced inhibition of migration and invasion, as revealed by the significant increases in these parameters compared to their values in the controls (Fig. 3D and E). Therefore, the upregulation of miR-155 regulates the inhibitory effects of PEG-IFN on HepG2 cell growth.
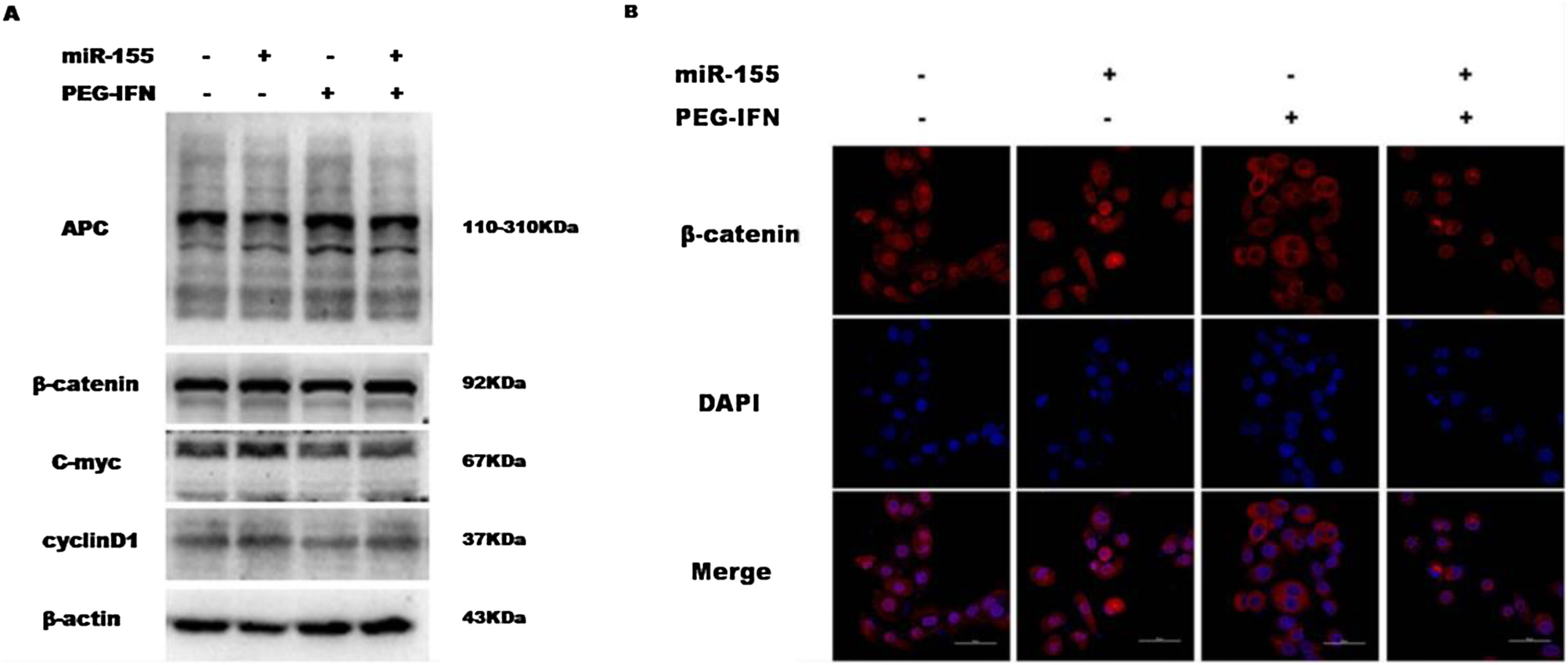
3.4Upregulation of miR-155 regulates the anti-proliferative effect of PEG-IFN through the Wnt/β-catenin pathwayThe upregulation of miR-155 promotes hepatocyte proliferation and tumourigenesis by activating Wnt signalling, while PEG-IFN exerts its anti-proliferative effect by inhibiting Wnt signalling. Therefore, this study also investigated whether miR-155 regulates the anti-proliferative effect of PEG-IFN on HepG2 cells through the Wnt/β-catenin pathway. As shown in Fig. 4A and B, miR-155 upregulation decreased the level of APC protein and increased the activity of the Wnt pathway by increasing the levels of nuclear β-catenin and downstream target genes (cyclin D1 and C-myc), regardless of any effect of the PEG-IFN treatment. In contrast, the downregulation of miR-155 by PEG-IFN was associated with an increase in APC levels and reduced the activity of the Wnt pathway by decreasing the levels of nuclear β-catenin and downstream target genes (cyclin D1 and C-myc). Based on these data, the upregulation of miR-155 regulates the inhibitory effects of PEG-IFN on HepG2 cell proliferation through the Wnt/β-catenin signalling pathway.
 After transfected with miR155 mimics, the cells were treated with 3600ng/ml PEG-IFN. miR-155 mimics significantly increased β-catenin, C-myc and Cyclin D1 protein expression and decreased APC protein expression in HepG2 cells with or without PEG-IFN treatment. (B) miR-155 mimics significantly increased β-catenin nuclear accumulation in HepG-2 cells with or without PEG-IFN treatment. β-Catenin signal was shown in red. Nuclei was shown in blue (DAPI). Scale bars: 50μm.")
Upregulation of miR-155 regulated PEG-IFN-mediated inhibition of cells proliferation through wnt/β-catenin signaling pathway. (A) After transfected with miR155 mimics, the cells were treated with 3600ng/ml PEG-IFN. miR-155 mimics significantly increased β-catenin, C-myc and Cyclin D1 protein expression and decreased APC protein expression in HepG2 cells with or without PEG-IFN treatment. (B) miR-155 mimics significantly increased β-catenin nuclear accumulation in HepG-2 cells with or without PEG-IFN treatment. β-Catenin signal was shown in red. Nuclei was shown in blue (DAPI). Scale bars: 50μm.
Sustained cell growth and proliferation, which are hallmarks of cancer, are responsible for cancer-related deaths by disrupting the balance of growth promotion and growth limitation. The inhibition of cancer cell proliferation and migration has been confirmed as a core component of tumour therapy [20]. An understanding of the molecular mechanisms will be crucial for the development of new therapeutic strategies to successfully address this challenge. In our study, PEG-IFN inhibited HepG2 cell proliferation through the Wnt/β-catenin pathway in a mechanism regulated by miR-155.
We investigated the role of miR-155, a well-established oncomiR, in HCC to identify the novel mechanism underlying the anti-proliferative effects of PEG-IFN [21]. In our study, miR-155 was expressed in HepG2 cells, and its expression was significantly decreased by PEG-IFN treatment in a dose-dependent manner, reaching the maximum decrease at 48h following treatment with 3600ng/mL PEG-IFN. Furthermore, PEG-IFN exerted potent inhibitory effects on HepG2 cell proliferation, migration and invasion, consistent with previous studies [22,23]. However, the precise regulatory mechanisms underlying the PEG-IFN-induced anti-proliferative effects on HepG2 cells and differential miR-155 expression remain unclear.
Notably, miR-155 has been reported to be an important regulator of tumourigenesis, as it is involved in mediating the proliferation and migration of HCC cells. The silencing of miR-155 significantly reduces the proliferation and migration of cancer cells, and conversely, miR-155 overexpression significantly enhances cancer cell proliferation and migration, indicating the importance of miR-155 in tumour growth [24,25]. In our present study, the overexpression of miR-155 in HepG2 cells reduced the effects of PEG-IFN on cell proliferation, migration and invasion, suggesting that miR-155 plays crucial roles in the PEG-IFN-induced anti-proliferative effect. Although miR-155 was related to the PEG-IFN-induced anti-proliferative effect, the precise regulatory mechanism has yet to be examined.
Since HCC is a heterogeneous and multi-step disease that is not successfully treated by targeting a single gene of interest, an understanding of the regulatory networks of many molecules will aid in the exploration of effective therapeutic methods [26]. The Wnt/β-catenin pathway is a signal transduction pathway with important roles in development and tumourigenesis. This pathway affects cell cycle progression by controlling the activation of genes associated with proliferation; therefore, this pathway controls the growth and proliferation of cells [27,28]. The tumour suppressor protein APC, a direct functional target of miR-155, was expressed in HepG2 cells and played a central role in regulating cell proliferation by targeting the proto-oncogene β-catenin. APC inhibition may affect downstream molecules in the Wnt/β-catenin pathway, leading to stimulation of the growth, invasion and metastasis of tumours. However, increased APC levels accelerate the nuclear export of β-catenin, which decreases the concentration of nuclear β-catenin and its availability to TCF, leading to the transcriptional repression of Wnt target genes and ultimately the inhibition of cellular proliferation [29,30]. In this study, the upregulation of miR-155 significantly decreased APC protein levels and increased the activity of the Wnt pathway in HepG2 cells treated with or without PEG-IFN. In contrast, the downregulation of miR-155 by PEG-IFN was associated with increased APC levels and reduced activity of the Wnt pathway. Therefore, miR-155 upregulation might regulate the inhibitory effects of PEG-IFN on HepG2 cell proliferation through the Wnt/β-catenin signalling pathway. The detailed mechanisms remain unclear, and further clarification is required.
In conclusion, PEG-IFN reduced the malignancy of HCC cells at least partially by downregulating miR-155. Further studies are needed to validate the clinical relevance of PEG-IFN.
AbbreviationsHCC hepatolcellular carcinoma interferon-α pegylated interferon-α microRNA 155 quantitative real-time PCR v-myc avian myelocytomatosis viral oncogene homolog adenomatous polyposis coli
Zhang Y, Guan YH designed the research; Zhang Y, Wang L, Xu J, Du JH performed the research; Zhang Y, Li XF analyzed the data; Zhang Y, Li XF wrote the paper.
Conflict of interestThe authors declare that they have no competing interest.
This work was supported by Liaoning province Natural Science Foundation Project (201602207) and Dalian Municipal Science and Technology Innovation Foundation Project (2018JI3SN117).



