





Background. Hepatitis C virus (HCV) is associated with a higher prevalence of steatosis compared to the general population. Aim. Our aim was to assess the impact of PNPLA3 rs738409 G-allele on steatosis in HCV patients.
Material and methods. We included 474 HCV patients treated with peginterferon plus ribavirin. PNPLA3 rs738409 was genotyped and patients were classified according to alleles and genotypes. Steatosis was detected in 46.4% (220/474). Fibrosis was assessed by Scheuer score. Gene expression was analyzed in Huh7.5 and Huh7 cells using Real Time-PCR.
Results. PNPLA3 allele-G was associated with steatosis [54.1% (126/233) vs. 39% (94/241)] (p = 0.0001). In HCV-1, allele-G was related to steatosis [50.6% (82/162) vs. 32.3% (53/164)] (p = 0.001), but did not in HCV-3 [61.9% (26/42) vs. 62% (31/50)] (p = 0.993). PNPLA3 allele-G was associated with steatosis in patients with IL28B-CT/TT [57.7% (82/142) vs. 37.1% (56/151)] (p = 0.0001), but did not in IL28B-CC [47.8% (43/90) vs. 42% (37/88)] (p = 0.442). Independent variables associated with steatosis were: PNPLA3 G-allele [O.R. 1.84 (CI95%: 1.06-3.21); p = 0.007], age [O.R. 1.04 (CI95%: 1.01-1.07); p = 0.017], HCV-genotype 3 [O.R. 2.46 (CI95%: 1.30-4.65); p = 0.006], HOMA > 4 [O.R. 2.72 (CI95%: 1.27-5.82); p = 0.010]. Since PNPLA3 RNA could not be detected on PBMC from HCV patients, an in vitro analysis was performed. Huh7.5 cells infected with JFH1 had a decreased PNPLA3 gene expression (fold inhibition = 3.2 ± 0.2), while Huh7 cells presented increased PNPLA3 gene expression (fold induction = 1.5 ± 0.2).
Conclusion. PNPLA3 allele-G modulated the development of steatosis, particularly in patients with HCV-1 and IL28B-CT/TT genotype, but was not associated with SVR. Metabolic but not viral steatosis seems to be PNPLA3 regulated. Gene interaction may result in differential PNPLA3 gene expression levels in HCV infection.
Liver steatosis is frequent among patients with chronic hepatitis C virus; its prevalence is higher than general population and individuals with other chronic liver diseases.1 It has been associated with poor sustained viral response (SVR) and more advanced hepatic fibrosis.2 Metabolic abnormalities, including overweight and type 2 diabetes mellitus, are thought to play an important role in the steatosis development in non-genotype 3 infected individuals, while hepatitis C virus (HCV) genotype 3-associated hepatic steatosis appears to be a result from a direct cytopathic effect.3 In 2008, Romeo, et al. ran an independent genome wide association study to identify genetic determinants of liver steatosis.4 A single genetic variant, I148M, encoding an isoleucine-to-methionine substitution at position 148 in the human patatin-like phospholipase domain containing 3 (PNPLA3) rs738409 C > G single nucleotide polymorphism (SNP) was identified as the strongest determinant of hepatocyte steatosis. Several candidate gene studies have demonstrated that PNPLA3 allele-G influences on liver fat accumulation. Additionally to NAFLD, PNPLA3 I148M sequence variant has been extensively associated with NASH,5 fibrosis progression,6 alcoholic liver disease7 and hepatocellular carcinoma.8 On the other hand, IL28B polymorphism CC has been related to higher SVR rate. However, its relationship with liver steatosis remains unclear, although suggested CC genotype could be related to lower steatosis degree.9 The primary aim of this study was to evaluate the association of PNPLA3 I148M variant with steatosis and fibrosis severity in a Spanish cohort of hepatitis C patients, while the second aim was to seek the interaction among genetic, metabolic and viral factors on steatosis development.
Material and MethodsIn this cross-sectional study, 474 consecutive patients with chronic hepatitis C infection, selected for peginterferon (PEG-IFN) and ribavirin (RBV) therapy, were enrolled from 14 Spanish Hospitals between October 2002 and January 2010. All patients followed standard treatment duration (24 or 48 week duration) according to HCV genotype. Patients received PEG-IFN-alpha (either Pegintron; Shering-Plough 1.5 mcg/kg/wk or Pegasys; Roche 180 mcg/wk) combined with RBV 1,000 mg/day if body weight was 75 kg or less, or 1,200 mg if body weight was greater than 75 kg. PEG-IFN and RBV dose modification followed standard criteria and procedures.10 Inclusion criteria were: patients > 18 years of age diagnosed as having chronic hepatitis C with HCV-RNA positive, who had criteria for commencing antiviral therapy in clinical practice; classified as genotype 1, 2, 3 or 4. Exclusion criterium was the presence of any coexisting chronic liver disease (including HIV and HBV infection). All patients completed the scheduled treatment.
All patients provided written informed consent for the collection and storage of peripheral blood mononuclear cells, as well as host DNA testing for research proposes consistent with the current study. The database for this analysis included clinical and demographic data extracted from the original clinical database, which included data about liver, kidney and metabolic profiles (particularly, glucemic and lipid metabolism). The study was approved by the Ethics Committee of each center and was conducted in accordance with the provisions of the declaration of Helsinki and good Clinical Practice Guidelines.
Viral load and HCV genotypingAt baseline, all patients had a quantitative measure of serum or plasma HCV-RNA performed by polymerase chain reaction assay using the COBAS AmpliPrep/COBAS TaqMan HCV Test (Roche Diagnostics GMBH, Mannheim) with a lower limit detection of 15 UI/mL. A qualitative or quantitative measurement of serum or plasma HCV-RNA was performed at week 4, 8, 12, 24 and 48 and follow-up weeks 4, 12, and 24. HCV genotyping was performed by reverse hybridization (Versant HCV® 2.0 Assay LiPA, Siemens) in all patients.
PNPLA3 genotypingThe rs738409 SNPs were analyzed using the StepOnePlus Real Time PCR System (Applied Biosystems, Foster City, USA) with a TaqMan SNP Genotyping Assay developed together with Applied Biosystems using published sequences from the NCBI Entrez SNP Database (www.ncbi.nlm. nih.gov/sites/entrez) (rs738409: 5’-AAGGAGGGA-TAAGGCCACTGTA-3’ as forward and 5’-CTT-TCACAGGCCTTGGTATGTTC-3’ as reverse primer).
IL28B genotypingThe genomic region associated with HCV treatment response lies on chromosome 19 and contains multiple SNPs in linkage disequilibrium around the IL28B gene. We selected the most strongly associated SNP, rs12979860, located 3-kb upstream of the IL28B gene, for genotyping in the cohorts by RTPCR using the LightCycler® 480 System (Roche Diagnostic).
Histological featuresAll of patients underwent to liver biopsy before therapy. Histologic evaluation was carried out by the same pathologist at each Hospital Center by Scheuer scoring:11
- •
F0: none portal fibrosis.
- •
F1: some-most portal fibrosis.
- •
F2: few bridging fibrosis.
- •
F3: many bridging fibrosis.
- •
F4: cirrhosis.
Steatosis was defined as > 5% of hepatocytes with macrovesicular steatosis.
Cell cultures and gene expression analysisHuh7.5 (rs12979860 genotype CT) and Huh7 (rs12979860 genotype CC) cells were grown in DMEM culture medium supplemented with 10% FBS, antibiotics, L-Glutamine and Non-Essential aminoacids. Cells were incubated at 37 oC, 5% CO2. Serum from patients infected by HCV-genotypes 1 and 3 with high viral load (> 107 IU/mL) were used for cell culture infection. Total RNA was extracted from cellular lysates using standard protocols (TRIsure™, BIOLINE, London, UK). JFH1 full genomic replicon was used to infect Huh7 and Huh7.5 cells, using 1 particle per cell. After infection, cells were incubated (37 oC, 5% CO2) for 48 h and then collected for total RNA isolation. We have performed the respective retro-transcription reactions using commercially available kits (Qiagen, Invitrogen, Carlsbad, CA, USA). Gene expression was analyzed by semi-quantitative real-time PCR using a illumina® Eco™ cycler. Primers for ACC (Acetyl-CoA Carboxylase), APOB (Apolipoprotein B), DGAT1 (Diacylglycerol O-Acyltransferase 1), DGAT2, FASN (Fatty Acid Synthase), LDLr (LDL cholesterol receptor), MTP (Microsomal triacylglycerol transfer protein), PPARG (Peroxisome Proliferator-activated Receptor Gamma), PNPLA3 (Patatin-like phospholipase domain containing 3) and SREBP (Sterol Regulatory Element-Binding Protein) genes were obtained from Qiagen (Hilden, Germany). GAPDH (Glyceraldehyde-3-phosphate Dehydrogenase) was used as a house-keeping gene.
Statistical analysisStatistical analyses and graphs were done with SPSS (19.0, SPSS Inc., Chicago, IL). All values are presented as means ± SD. Comparisons between groups were made using the Mann-Whitney U test, the Student t-test or ANOVA for continuous variables, and the Chi-square or the Fisher exact probability test for categorical data. Variables that showed significance p < 0.10 in univariate analysis were entered into backward logistic regression analysis. The multivariate models were constructed sequentially with variables entered one at a time and a significance level of 0.05 was used to remove them from the model, except age and sex, which were included in all the models.
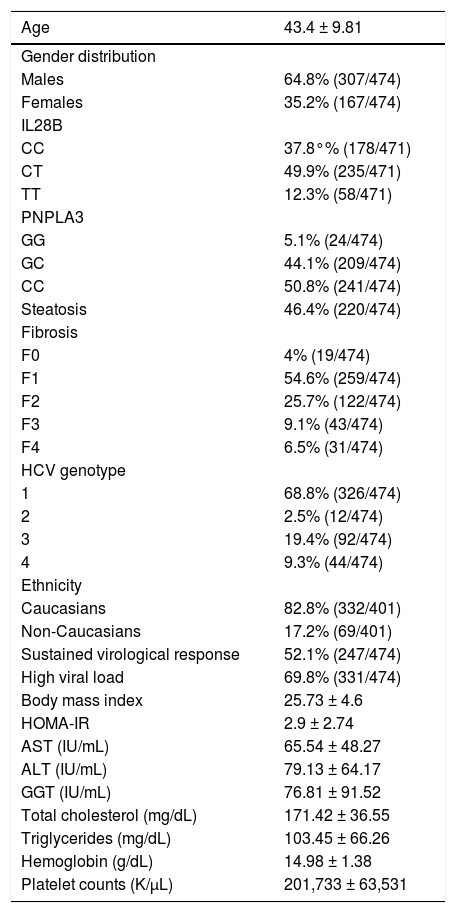
ResultsBaseline epidemiological and biochemical features of overall cohort are shown in table 1. Gender distribution was 64.8% (307/474) males and 35.2% (167/474) females, with a mean age of 43.4 ± 9.81 years of age.
Baseline characteristics of overall cohort.
| Age | 43.4 ± 9.81 |
|---|---|
| Gender distribution | |
| Males | 64.8% (307/474) |
| Females | 35.2% (167/474) |
| IL28B | |
| CC | 37.8°% (178/471) |
| CT | 49.9% (235/471) |
| TT | 12.3% (58/471) |
| PNPLA3 | |
| GG | 5.1% (24/474) |
| GC | 44.1% (209/474) |
| CC | 50.8% (241/474) |
| Steatosis | 46.4% (220/474) |
| Fibrosis | |
| F0 | 4% (19/474) |
| F1 | 54.6% (259/474) |
| F2 | 25.7% (122/474) |
| F3 | 9.1% (43/474) |
| F4 | 6.5% (31/474) |
| HCV genotype | |
| 1 | 68.8% (326/474) |
| 2 | 2.5% (12/474) |
| 3 | 19.4% (92/474) |
| 4 | 9.3% (44/474) |
| Ethnicity | |
| Caucasians | 82.8% (332/401) |
| Non-Caucasians | 17.2% (69/401) |
| Sustained virological response | 52.1% (247/474) |
| High viral load | 69.8% (331/474) |
| Body mass index | 25.73 ± 4.6 |
| HOMA-IR | 2.9 ± 2.74 |
| AST (IU/mL) | 65.54 ± 48.27 |
| ALT (IU/mL) | 79.13 ± 64.17 |
| GGT (IU/mL) | 76.81 ± 91.52 |
| Total cholesterol (mg/dL) | 171.42 ± 36.55 |
| Triglycerides (mg/dL) | 103.45 ± 66.26 |
| Hemoglobin (g/dL) | 14.98 ± 1.38 |
| Platelet counts (K/μL) | 201,733 ± 63,531 |
HCV-genotype distribution was:
- •
68.8% (326/474) patients with genotype 1.
- •
2.5% (12/474) patients with genotype 2.
- •
19.4% (92/474) patients with genotype 3; and
- •
9.3% (44/474) patients with genotype 4.
PNPLA3 polymorphism was genotyped:
- •
GG 5.1% (24/474).
- •
GC 44.1% (209/474).
- •
CC 50.8% (241/474); G allele 49.2% (233/474) vs. 50.8% (241/474) non-G alelle.
IL28B polymorphism was analyzed:
- •
CC 37.8% (178/471).
- •
CT 49.9% (235/471), and
- •
TT 12.3% (58/471); IL28B-CC 37.8% (178/471) vs. 62.2% (293/471) IL28B-CT/TT.
Patients showing > 800.000 IU/mL [69.8% (331/ 474)] were considered as high baseline viral load. Steatosis was observed in 46.4% (220/474) of patients.
Fibrosis stage was measured:
- •
4% (19/474) F0.
- •
54.6% (259/474) F1.
- •
25.7% (122/474) F2.
- •
9.1% (43/474) F3; and
- •
6.5% (31/474) F4.
According to ethnicity, 82.8% (332/401) were Caucasians and 17.2% (69/401) were from other ethnicities.
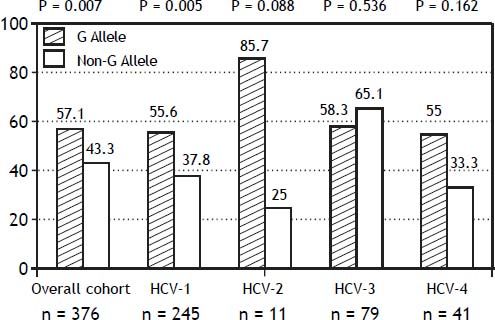
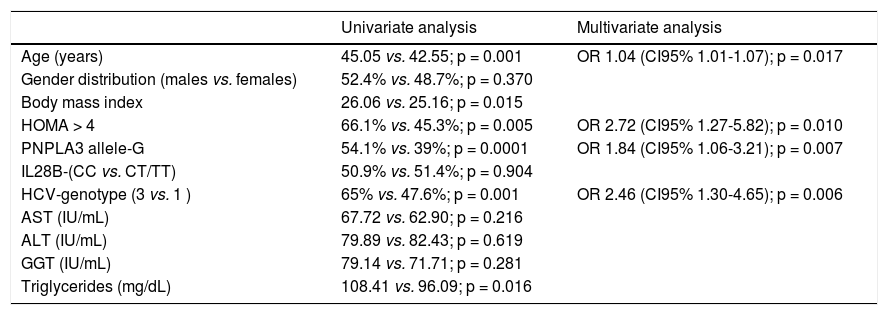
The PNPLA3 allele-G did not show steatosis in patients with IL28B-CC [47.8% (43/90) vs. 42% (37/ 88) in PNPLA3 non-allele-G] (p = 0.442). However, in patients with IL28B-CT/TT steatosis was more prevalent in PNPLA3 allele-G (57.7%; 82/142) than in non-allele-G (37.1%; 56/151) (p = 0.0001) (Figure 1). In the overall cohort (all HCV-genotypes), steatosis was present in 54.1% (126/233) in patients with PNPLA3 allele-G, while it was detected in 39% (94/241) in non-allele-G (p = 0.0001). In HCV-genotype 1 (n = 326), PNPLA3 allele-G influenced on the presence of steatosis (50.6%; 82/162) vs. non-allele-G (32.3%; 53/164) (p = 0.001). PNPLA3 allele-G was associated with steatosis in non-3 genotype (52.4%; 100/191 vs. 33%; 63/191) (p = 0.0001). In contrast, allele-G did not show any impact on steatosis in HCV-genotype 3 (61.9%; 26/42 vs. 62%; 31/50) (p = 0.993) (Figure 2). On the other hand, HOMA was higher in patients with steatosis (3.92 vs. 2.49; p = 0.013), as well as body mass index (26.06 vs. 25.16; p = 0.015). In multivariate analysis, variables independently associated with steatosis were: PNPLA3 allele-G [O.R. 1.84 (CI95%: 1.06-3.21); p = 0.007], age [O.R. 1.04 (CI95%: 1.01-1.07); p = 0.017], HCV-genotype 3 [O.R. 2.46 (CI95%: 1.30-4.65); p = 0.006] and HOMA > 4 [O.R. 2.72 (CI95%: 1.27-5.82); p = 0.010] (Table 2).
, according to PNPLA3 allele, by IL28B genotype distribution.")
, according to PNPLA3 allele, by HCV-genotype distribution. HCV: hepatitis C virus.")
Univariate and multivariate analysis according to steatosis.
| Univariate analysis | Multivariate analysis | |
|---|---|---|
| Age (years) | 45.05 vs. 42.55; p = 0.001 | OR 1.04 (CI95% 1.01-1.07); p = 0.017 |
| Gender distribution (males vs. females) | 52.4% vs. 48.7%; p = 0.370 | |
| Body mass index | 26.06 vs. 25.16; p = 0.015 | |
| HOMA > 4 | 66.1% vs. 45.3%; p = 0.005 | OR 2.72 (CI95% 1.27-5.82); p = 0.010 |
| PNPLA3 allele-G | 54.1% vs. 39%; p = 0.0001 | OR 1.84 (CI95% 1.06-3.21); p = 0.007 |
| IL28B-(CC vs. CT/TT) | 50.9% vs. 51.4%; p = 0.904 | |
| HCV-genotype (3 vs. 1 ) | 65% vs. 47.6%; p = 0.001 | OR 2.46 (CI95% 1.30-4.65); p = 0.006 |
| AST (IU/mL) | 67.72 vs. 62.90; p = 0.216 | |
| ALT (IU/mL) | 79.89 vs. 82.43; p = 0.619 | |
| GGT (IU/mL) | 79.14 vs. 71.71; p = 0.281 | |
| Triglycerides (mg/dL) | 108.41 vs. 96.09; p = 0.016 |
In contrast, PNPLA3 did not influence on fibrosis severity neither SVR when patients were stratified by viral or IL28B genotypes (data not shown). Indeed, PNPLA3 allele-G achieved SVR in 50.2% (117/233), while non-allele-G in 53.9% (130/241) (p = 0.417).
In vitro analysis (Figure 3) showed that Huh7.5 cells (CT genotype) infected by HCV-genotype 1 had significantly increased gene expression (> 1.5 fold induction) for DGAT1 and DGAT2 genes. In Huh7 cells (CC genotype) APOB, DGAT1, DGAT2, LDLR and SREBP were found induced. Infection by HCV-genotype 3 promoted increased gene expression of |ACC, FASN and MTP in Huh7.5 while DGAT1, DGAT1, FASN and MTP were significantly induced in Huh7 cells. PNPLA3 gene expression was also analyzed in these cells (Figura 3C): in Huh7.5 (IL28B CT genotype) cells infected with JFH1, a decreased gene expression was observed (fold inhibition = 3.2 ± 0.2). By contrary, in Huh7 cells harboring IL28B CC genotype, the opposite effect was detected: PNPLA3 gene expression was induced (fold induction = 1.5 ± 0.2).
 in Huh7.5 (IL28B CT genotype) (A) and Huh7 (IL28B CC genotype) (B) cells infected with JFH1 particles. Lipid metabolism related gene expression was analyzed by semi-quantitative real time-PCR (see methods for details). C. PNPLA3 gene expression in Huh7 and Huh7.5 cells infected with JFH1 particles. Non-infected cell cultures are used as control. Data shown are mean values from three independent experiments. G1: genotype 1. G3: genotype 3.")
Gene expression (fold induction) in Huh7.5 (IL28B CT genotype) (A) and Huh7 (IL28B CC genotype) (B) cells infected with JFH1 particles. Lipid metabolism related gene expression was analyzed by semi-quantitative real time-PCR (see methods for details). C. PNPLA3 gene expression in Huh7 and Huh7.5 cells infected with JFH1 particles. Non-infected cell cultures are used as control. Data shown are mean values from three independent experiments. G1: genotype 1. G3: genotype 3.
We reported an association between PNPLA3 rs738409 allele-G and liver steatosis in a large cohort of chronic hepatitis C patients. Indeed, the impact of PNPLA3 polymorphisms on steatosis depends on viral and IL28B genotypes. Steatosis is known to negatively impact on the natural history of HCV infection as it accelerates the progression to cirrhosis. Metabolic, enviromental, genetic and viral factors have been implicated on steatosis development. However, the weight of each of them in a determinate patient remains elusive. Patients with PNPLA3 allele-G, overweight, HOMA > 4 and viral genotype 3a were independently associated with steatosis. PNPLA3 allele-G was associated with steatosis in patients infected by HCV-genotype 1, showed a trend in genotype 2 and genotype 4, but was not related to steatosis in genotype 3a. A link between this gene and steatosis was reported by Trépo, et al., but genotype 3 patients were not included.12 Our data confirm previous results reported by Valenti, et al.13 and Cai, et al.14 that evaluated the effect of PNPLA3 on liver fat and fibrosis in HCV patients. The rs738409 GG genotype was closely associated with steatosis across all viral genotypes except for genotype 3a. However, Rembeck, et al., did not find differences in the prevalence of steatosis between HCV-genotypes 2 and 3 in patients with PNPLA3 rs738409 allele-G, in spite of HCV-genotype 2 showed an increased insulin resistance.15 These data could indicate two types of liver steatosis in chronic hepatitis C patients:
- •
Metabolic steatosis promoted by genetic factors (PNPLA3 allele-G) in HCV-genotypes 1, 2 and 4.
- •
Viral steatosis related to HCV-genotype 3.
On the other hand, PNPLA3 polymorphisms influenced on steatosis depending on IL28B genotype. PNPLA3 allele-G increased the risk of steatosis in patients with IL28B-CT/TT but did not in genotype IL28B-CC. The rational for considering the effect of PNPLA3 on steatosis according to IL28B genotype was to identify some factor which explains the higher presence of steatosis in IL28B-CT/TT patients. In fact, Valenti, et al. observed that IL28B-CC protected from steatosis in PNPLA-GG patients, but did not in those negative for the PNPLA3 G variant at risk, in non genotype-3 patients.16 Tillman, et al. demonstrated that the presence of steatosis was lower in IL28B-CC compared to non-CC patients in two independent HCV-genotype 1 cohorts.17 Recently, similar results have been documented by Agundez, et al.18 Therefore, the different effect of PNPLA3 alleleG according to IL28B genotype could explain, at least in part, the higher prevalence of steatosis in IL28B-CT/TT patients. Our in vitro results showed that cells harboring IL28B-CC genotype induced greater gene expression than IL28B-CT genotype cells in HCV genotype 3 infection, in particular for MTP (Figure 3). As pointed out by Mirandola, et al.19 there is a complex relationship between MTP and HCV: since MTP is essential for the assembly and secretion of HCV, MTP expression should be upregulated to facilitate its propagation during early infection stages, although MTP mRNA levels are reduced in chronic HCV patients independently of HCV genotype.20 Pharmacologic inhibition of MTP has been proposed as a potential antiviral strategy for HCV.21
PNPLA3 is a member of the patatin-like phospholipase family which encodes a transmembrane protein expressed in stellate cells but more prominently in hepatocytes, where interacts with lipids. The mechanism by which variation in PNPLA3 affects liver triglyceride content is not completely known. The PNPLA3 rs738409 G-allele was suggested to impair triglyceride hydrolysis in hepatocytes and favour the increase of triglycerides accumulation.22 Both Huh7.5 (IL28B CT genotype) and Huh7 (IL28B CC genotype), harbor the GG genotype for rs738409 polymorphism, being a good system for studying gene interaction with IL28B genotypes. Our results demonstrated that JFH1 increased PNPLA3 expression in favorable IL28B genotype cells. Since lower PNPLA3 gene expression and activity are associated to steatosis development, the interaction between IL28B, PNPLA3 polymorphism and HCV infection could explain the grade of steatosis and sustained viral response.
On the other hand, it has been suggested that accumulation of fat in the hepatocyte cytoplasm in form of large lipid droplets favors virion assembly. Otherwise, DGAT1 and DGAT2 enzymes catalyze the final step in triglyceride biosynthesis and are essential in lipid droplet biogenesis, especially DGAT1.23 Interestingly, DGAT1 (required for efficient viral particle formation24) and DGAT2 genes were found induced in genotype 1 compared to genotype 3 in cells with IL28B-CT genotype. This effect was not observed in IL28B-CC cells, indicating a host-virus genetic interaction.
PNPLA3 polymorphisms were not associated with sustained virological response. This fact confirms data from previous studies that showed PNP-LA3 rs738409 allele-G did not influence on treatment response.25,26 Nevertheless, PNPLA3 polymorphisms have been associated with increased risk of hepatocellular carcinoma in alcoholic cirrhosis.27 HCC is developed in some cirrhotic patients despite SVR.28 If PNPLA3 genotype, through promoting steatosis, could be implicated on this process requires further studies.
This study has some limitations. First, the design of the study (cross-sectional) is not the appropiate method to stablish cause-effect relationships. However, most of data are consistent with hypothesis about PNPLA3. Second, the number of patients with HCV-genotype non-1 was limited, particularly HCV-genotypes 2 and 4 (HCV-1 genotype is the most prevalent genotype in Spain), which could underestimated the effect of PNPLA3 on liver steatosis in these HCV-genotypes.
In conclusion, PNPLA3 rs738409 allele-G is shown as a crucial predictive factor of liver steatosis in HCV-genotype 1 but did not in genotype 3. These data demonstrate that PNPLA3 rs738409 allele-G generates metabolic steatosis, while steatosis in HCV-genotype 3 could be promoted by direct viral effect. Furthermore, PNPLA3 allele-G has a stronger effect in IL28B-CT/TT genotype than in IL28B-CC. Indeed, lipids-related gene expression was modulated by viral genotype according to IL28B background in vitro. These findings support that steatosis is the final result of the interaction between host and viral factors.
Abbreviations- •
APOB: apolipoprotein B.
- •
ACC: acetyl-CoA carboxylase.
- •
DGAT: diglyceride acyltransferase.
- •
FASN: fatty acid synthase.
- •
HCC: hepatocellular carcinoma.
- •
HCV: hepatitis C virus.
- •
LDLr: LDL receptor.
- •
MTP: microsomal triaglyceride transfer protein.
- •
NAFLD: non-alcoholic fatty liver disease.
- •
NASH: non-alcoholic steatohepatitis.
- •
PEG-IFN: peginterferon.
- •
PNPLA3: patatin-like phospholipase domain containing 3.
- •
PPARG: peroxisome proliferator-activated receptor gamma.
- •
RBV: ribavirin.
- •
SNP: single nucleotide polymorphism.
- •
SREBP: sterol regulatory element-binding protein.
- •
SVR: sustained viral response.
- •
Planning and conducting the study: Javier Ampuero, Manuel Romero-Gomez.
- •
Drafting the manuscript: Javier Ampuero, Jose Antonio del Campo, Manuel Romero-Gomez.
- •
Interpreting data: Javier Ampuero, Jose Antonio del Campo, Manuel Romero-Gomez.
- •
Performing in vitro studies: Jose Antonio del Campo, Lourdes Rojas.
- •
Collecting data: Jose Raúl García-Lozano; Ricard Solá; Raúl Andrade; José Antonio Pons; Jose María Navarro; Jose Luis Calleja; María Buti; María Francisca González-Escribano; Xavier Forns, Moisés Diago, Javier García-Samaniego.
Guarantor of the article: Manuel Romero-Gomez.
Financial SupportNone.
Potential Competing InterestNone.



