



Background and aim. The HBV covalently closed circular DNA (cccDNA) is organized into a minichromosome in the nuclei of infected hepatocytes through interactions with histone and nonhistone proteins. Retinoid X receptor a (RXRa), a liver-enriched nu-clear receptor, participates in regulation of HBV replication and transcription through modulation of HBV enhancer 1 and core promoter activity.
Material and methods. This study investigated RXRa involvement in HBV cccDNA epigenetic modifications. Quantitative cccDNA chromatin immunoprecipitation (ChIP) was applied to study the recruitment of RXRa, histones, and chromatin-modifying enzymes to HBV minichromosome in HepG2 cells after transfection of the linear HBV genome.
Results. RXRa was found to directly bind to HBV cccDNA; recruitment of RXRa to HBV minichromosome paralleled HBV replication, histone recruitment, and histone acetylation in HBVcccDNA. Moreover, RXRa overexpression or knock-down significantly increased or impaired the recruitment of the p300 acetyltransferase to cccDNAminichromosome.
Conclusions. Our results confirmed the regulation of RXRa on HBV replication in vitro and demonstrated the modulation of RXRa on HBV cccDNA epigenetics. These findings provide a profound theoretical and experimental basis for late-model antiviral treatment acting on the HBV cccDNA and minichromosome.
Chronic hepatitis B virus (HBV) infection is a major health problem worldwide and frequently results in liver cirrhosis or hepatocellular carcinoma.1 HBV is an enveloped and partially double-stranded small DNA virus. Following infection, the HBV 3.2-kb open circular duplex (OC) DNA genome is converted into a covalently closed circular DNA (cccDNA) in the nucleus of the infected hepatocytes, and serves as a template for the transcription of all viral messenger RNAs and 3.5-kb pre-genomic RNA (pgRNA).2,3 The cccDNA has been proven as the basis of persistence and recurrence of HBV infection because of its long half-life and relative resistance to therapeutic regimens.2,4,5
HBV cccDNA has been shown to be organized into a viral minichromosome in the nuclei of infected hepatocytes through interactions with histone and nonhistone proteins.6 Using cccDNA chromatin immunoprecipitation (ChIP) assays, previous studies have shown that HBV replication is regulated by the acetylation status of cccD-NA-bound H3/H4 histones, and the HBV X protein (HBx) influences cccDNA epigenetic changes by modulating recruitment of chromatin-modifying enzymes onto the viral minichromosome.7-9
HBV infection displays a tissue tropism that is restricted primarily to the liver.10,11 Previous studies have demonstrated that liver-enriched transcription factors such as hepatocyte nuclear factor 4α (HNF4α), retinoid X receptor a (RXRα), and peroxisome proliferator-activated receptor α (PPARα) are involved in HBV infection hepatotropismand regulation of HBV replication and transcription.12-15 Among these, RXRα has been identified as an important transcription factor in regulating HBV pgRNA synthesis through binding to HBV promoter regulatory elements, such as HBV enhancer I.13,14,16
In most cases, the binding of transcription factors to the regulating elements in target genes may be accompanied by local chromatin remodeling.17,18 We hypothesize that RXRα, like HBx, may be involved in cccDNA epigenetic modifications and, thereby, regulate HBV replication and transcription. In this study, we investigated the recruitment of RXRα onto HBV cccDNA and its ability for regulation of viral minichromosome remodeling in HBV replicating cells.
Material and MethodsTransient transfection of linear HBV DNA genomes and RXRa vectorsFull-length linear HBV DNA monomers were re-leased from the pUC19-HBV1.0 plasmids by Sap I (Thermo Scientific, Carlsbad, CA, USA) digestion as previously described.8 The human RXRα expression vector (pRS-hRXRα) was provided by Dr. Hong Tang (West China Medical School, Sichuan University, China). Human RXRα shRNA vector, containing three siRNA oligos and one negative control oligo, was purchased from Invitrogen Service. The most effective silencing shRNA plasmid was selected for subsequent experiments. HepG2 cells were split and seeded at a density of 1 x 106 cells in 10-cm diameter Petri dishes (Corning, Tewksbury, MA, USA) the day before transfections. Vectors and linear HBV monomers were transfected alone or cotransfected using X-treme GENE HP DNA transfection reagent (Roche, Mannheim, Germany). All-trans retinoic acid at 1 μM was used to activate the nuclear receptor RXRα.
HBV antigen quantificationHepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) in HepG2 culture supernatants were quantified by an enzyme-linked immunosorbent assay (ELISA) kit (KeHua, Shanghai, China) at different time points after transfection with linear HBV monomers or cotransfection with pRS-hRXRα or RXRα shRNA plasmid. Absorbance was detected at 450 nm by a microplate spectrophotometer and the final results were expressed as optical density (OD) values.
Southern blotting of cytoplasmic HBV DNAHepG2 cells were harvested at 48 h after transfection and resuspended in lysis buffer (10 mMTris-HCl pH8.0, 1 mM EDTA, 1%NP40, and 2% sucrose). PEG 8000 was added to the supernatants and the mixture was incubated on ice. After centrifugation, the precipitates were adjusted to 40 U/mL DNase I solution (Promega, Madison, WI, USA) and incubated at 37 °C. The mixture was incubated with proteinase K at 50 °C overnight followed by phenolchloroform extraction. Southern blotting was performed as described previously, with minor modifications.8,9 Briefly, DNA was blotted onto positively charged nylon membranes and hybridized with DIG (digoxigenin)-labeled gel-purified full-length HBV fragment. The probe was generated with a DIG High Prime DNA Labeling and Detection Starter Kit II (Roche, Basel, Switzerland).
Real-time PCR quantification of cytoplasmic HBV DNATotal cytoplasmic HBV DNA was extracted from HepG2 cells at 12, 24, 48, and 72 h after transfections and quantified by real-time PCR as described previously,8 using the following primers and probe: forward, 5’-AGAAACAACACATAGCGCCTCAT-3’, reverse, 5’-TGCCCCATGCTGTAGATCTTG-3’, and TaqMan probe, 5’-FAM-TGTG GGTCACCATATTCTTGGG-TAMER-3’. The plasmid pU19-HBV1.0 was serially diluted 10-fold as the standard for real-time PCR. Final results were expressed as the number of DNA copies per cell.
HBV cccDNA and pgRNA quantificationTotal RNA was extracted from HepG2 cells using RNAiso Plus (Takara, Japan). For pgRNA analysis, 1 μg of DNase-treated RNA was reverse transcribed and then 2 μL of each cDNA was quantified by a Light-Cycler PCR system (Roche, Basel, Switzerland) using the following pgRNA-specific primers and probes: forward, 5’-GCCT-TAGAGTCTCCTGAGCA-3’, reverse, 5’-GAGGGAGT-TCTTCTTCTAG G-3’, FRET hybridization probes, 5’-AGTGTGGATTCGCACTCCTCCAGC-FL-3’, and Red640-5’ATAGACCACCAAATGCCCCTATCTTAT-CAAC-3’; h-GAPDH was used to normalize the RNA samples.
HBV cccDNA were isolated and analyzed by a minor modification of a previously described protocol.8,9 Briefly, the precipitates of post-transfection HepG2 cells lysis were treated with nuclei lysis buffer (6% SDS, 100 mmol NaOH). The extracted HBV DNA was treated with Plasmid-Safe ATP-Dependent DNase (Epicentre Biotechnologies, Madison, Wisconsin) to degrade the contaminating HBV OC species. HBV cccDNA was quantified by Light-Cycler 480 Real-Time PCR System (Roche, Basel, Switzerland) using specific primers, 3’-fluorescein (FL)-labeled probe and 5’-Red640 (R640)-labeled probe, as described previously.8,9 Serial dilutions of pUC19-HBV1.0 plasmid were used as quantification standards.
Real-time PCR quantification of RXRa expressionTotal RNA was extracted from HepG2 cells after transfections and 1 μg RNA was used for cDNA synthesis using PrimeScript RT reagent kit with gDNA Eraser (Takara, Dalian, China), and followed by SYBR Green real-time PCR quantification by BioRad CFX96 real-time thermo-cycler (BioRad, Foster City, CA, USA). The relative RXRa mRNA expression levels were estimated using the ΔΔCT method with normalization to h-GAPDH RNA. The RXRa PCR primers were 5’-ACGAGAATGAGGT-GGAGTCG-3’ (sense), 5’-ATGTTGGTGACAG-GGTCGTT-3’ (antisense).
Western blotting analysis of RXRa expressionPost-transfection HepG2 cells were lysed in RIPA buffer containing 1 mM PMSF. Protein lysates were separated with 10% SDS-PAGE gel and transferred to a PVDF membrane and incubated with primary antibodies specific to RXRα (Millipore, Cat.# MAB5478: mouse monoclonal IgG) followed by incubation with its respective secondary goat anti-mouse antibody. Immunoreactive proteins were visualized with the enhanced chemiluminescence reagent (ECL Kit, Amersham Bio-science, Little Chalfont, Buckinghamshire, UK) and quantified using Western Workflow Complete System (BioRad, Foster City, CA, USA). The values were normalized to the intensity levels of β-actin.
Chromatin immunoprecipitation assaysChromatin immunoprecipitation (ChIP) assays were performed with EZ-ChIP Chromatin Immunoprecipitation Kit (Millipore, Germany) according to the manufacturer's recommendations, with minor modifications. Briefly, 300-600-bp cross-linked chromatin fragments were immunoprecipitated using antibodies specific to H4 (Cat.#17-10047: rabbit monoclonal IgG recognizing unmodified histone H4, Millipore), AcH4 (Cat.#06-598: rabbit polyclonal IgG recognizing histone H4 which is tet-ra-acetylated at lysine 5, 8, 12, and 16, Millipore), AcH3 (Cat.#07-353: rabbit polyclonal IgG recognizing histone H3 which is acetylated at lysine 14, Millipore), RXRα (Cat. #MAB5478: mouse monoclonal IgG, Millipore), p300 (Cat.#sc-585:rabbit polyclonal IgG, Santa Cruz Bio-technology) and histone deacetylase complex 1 (HDAC1;Cat. #06-720: rabbit polyclonal IgG, Millipore). Immunoprecipitations with relevant nonspecific immunoglobulins (normal rabbit IgG, Millipore) and anti-RNA Ploy II (Cat.#17-620: mouse monoclonal IgG1, Millipore) were included as a negative and positive control. After reverse cross-linking, immunoprecipitated chromatins were treated with Plasmid-Safe ATP-Dependent DNase (Gene Target Solutions Pty Ltd, Dural, NSW, Australia) and analyzed by real-time PCR for quantification of immunoprecipitated HBV cccDNA as described earlier. The final results were expressed as a percentage of inputs.
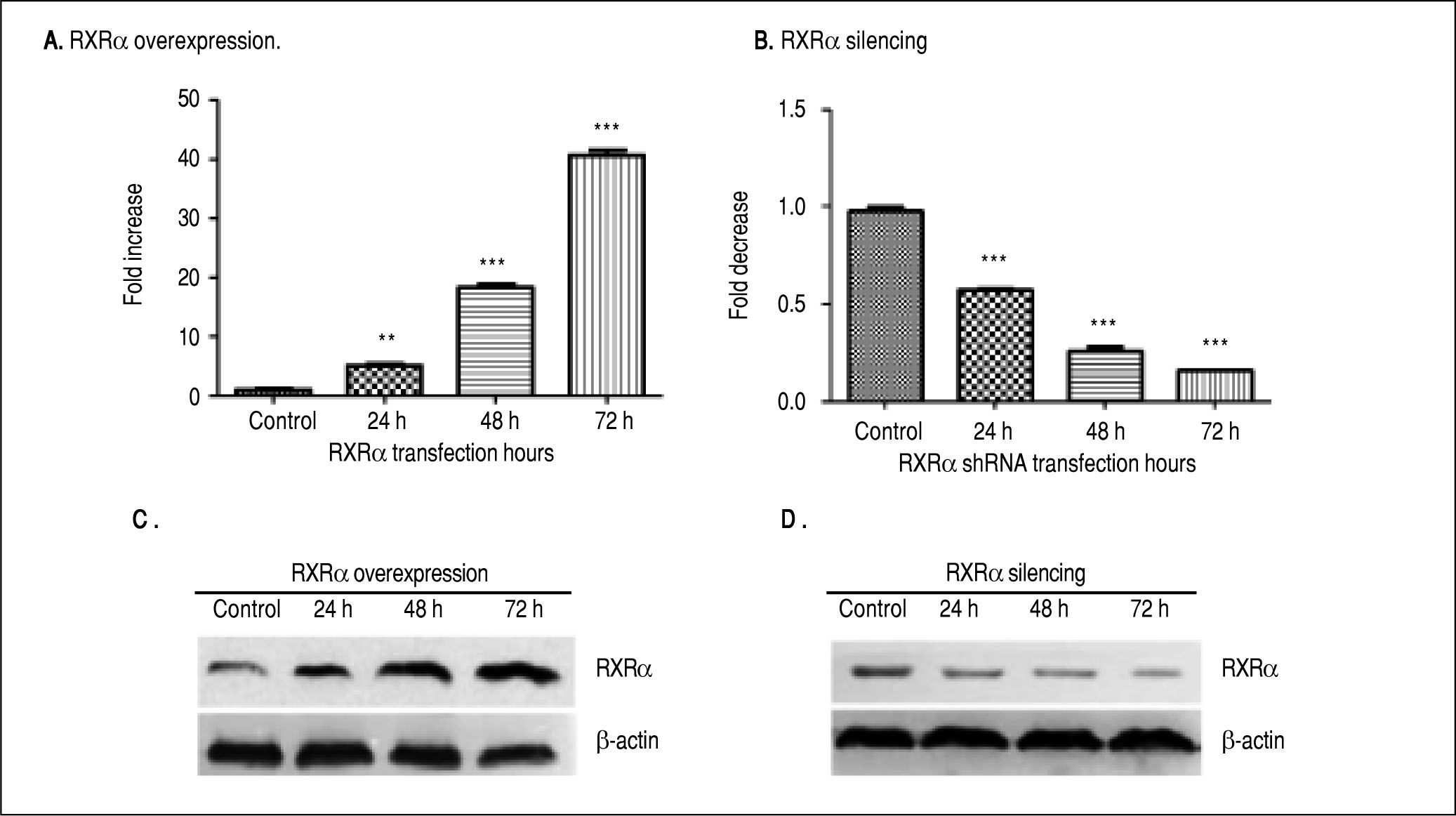
ResultsRXRα regulates HBV replication and transcription in HepG2 cellsHepG2 cells were transfected with RXRα mammalian expression or shRNA vectors. As shown infigure 1 A and 1B, RXRα overexpression or silencing significantly increased or decreased the RXRα mRNA levels in HepG2 cells, starting from 24 h and peaking at 72 h after transfection. RXRα protein levels followed the same pattern as mRNA after transfection of RXRa expression or shRNA (Figures 1C and 1D).
 or RXRα shRNA vectors (B). The RXRa mRNA levels were normalized by GAPDH. The indicated numbers in A and B are fold changes compared to HepG2 cells transfected with control pRS or negative shRNA vectors. * p < 0.05, ** p < 0.01, *** p < 0.001 (One-way ANOVA). C, D. Western blot detection of RXRv protein levels in HepG2 cells after transfection with pRS-hRXRα (C) or RXRα shRNA vectors (D) at the indicated time points. The experiment was repeated thrice and results of a representative assay shown.")
RXRα overexpression and knock down in HepG2 cells. A, B. Real-time PCR quantification of RXRα mRNA levels in HepG2 cells transfected with pRS-hRXRα (A) or RXRα shRNA vectors (B). The RXRa mRNA levels were normalized by GAPDH. The indicated numbers in A and B are fold changes compared to HepG2 cells transfected with control pRS or negative shRNA vectors. * p < 0.05, ** p < 0.01, *** p < 0.001 (One-way ANOVA). C, D. Western blot detection of RXRv protein levels in HepG2 cells after transfection with pRS-hRXRα (C) or RXRα shRNA vectors (D) at the indicated time points. The experiment was repeated thrice and results of a representative assay shown.
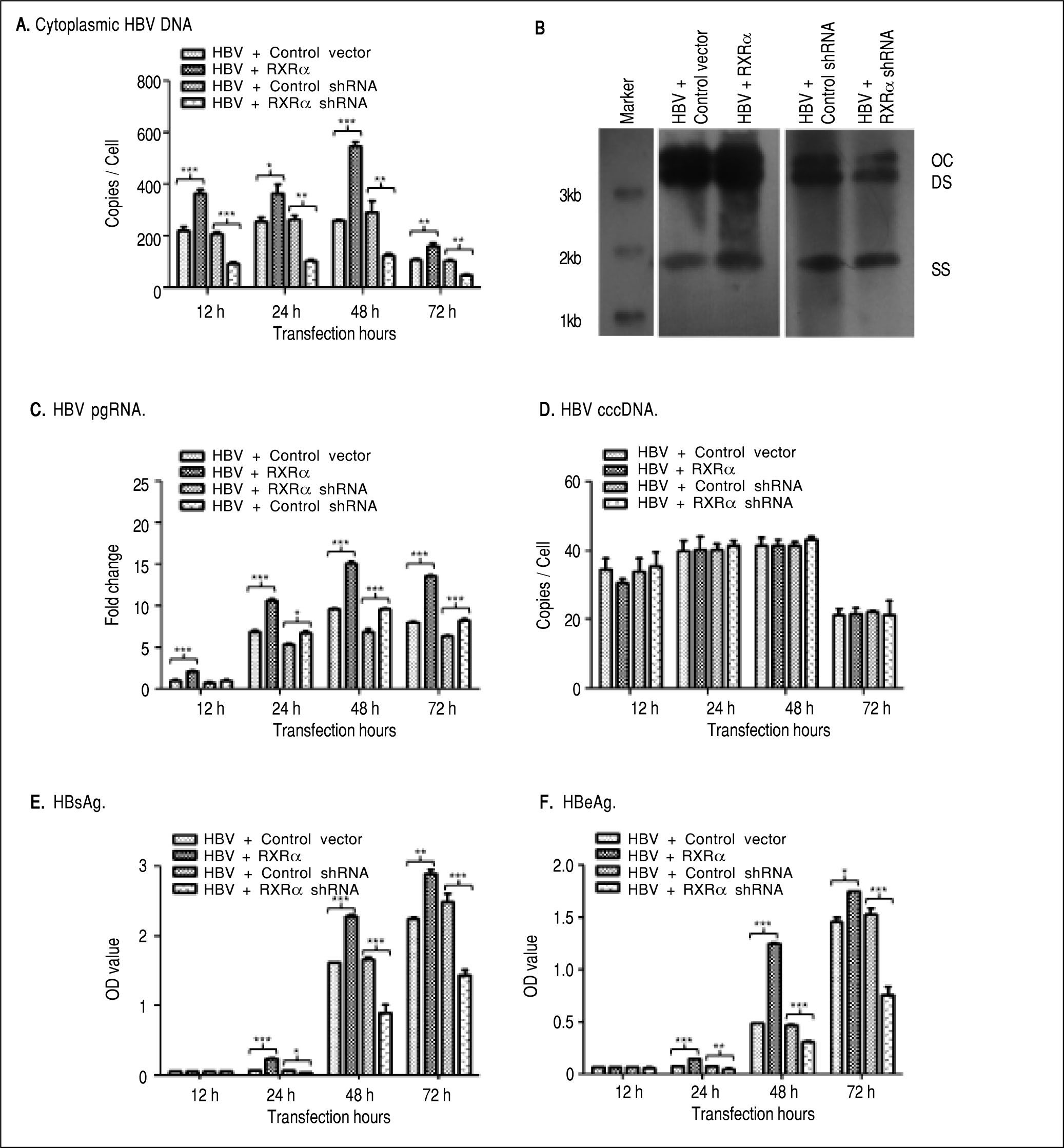
To determine whether RXRα directly modulates HBV replication and transcription, linear HBV DNA genomes were transfected or co-transfected with RXRa expression or shRNA vector into HepG2 cells. Cytoplasmic HBV DNA and pgRNA, nuclear cccDNA, and supernatant HB-sAg and HBeAg levels were determined by real-time PCR and ELISA, respectively. As shown infigure 2, after transfection of linear HBV DNA monomers, cytoplasmic HBV DNA, pgRNA, and nuclear cccDNA molecules were detected at 12 h, reached a peak at 48 h and declined at 72 h (Figures 2A, 2C, and 2D, respectively); the secreted HB-sAg and HBeAg were found elevated at 24 h and peaked at 72 h (Figure 2E and 2F). Interestingly, RXRα overexpression significantly increased the levels of all viral replication and transcription markers mentioned earlier at each time point, except for HBV cccDNA, and the same peaking patterns were observed as in transfection with linear HBV DNA alone (Figures 2A-2F). On the contrary, co-transfection with linear HBV DNA and RXRα shRNA vector dramatically reduced viral replication and transcription markers in HepG2 cells (Figures 2A-2F). Cytoplasmic HBV DNA accumulation after 48 h of co-transfection in HepG2 was confirmed on southern blotting analysis (Figure 2B). These results demonstrate that RXRα directly regulates HBV replication and transcription.
, pRS-hRXRa (HBV + RXRα group), negative shRNA (HBV + Control shRNA) or RXRα shRNA vectors (HBV + RXRα shRNA), respectively. A. Real-time PCR quantification of cytoplasmic HBV DNA in post-transfection HepG2 cells. Results are expressed as the number of cytoplasmic DNA copies per cell. B. Southern blot analysis of cytoplasmic HBV replicative intermediates in HepG2 cells after 48 h transfection. The experiment was repeated thrice, and results of a representative assay shown. OC, open circular duplex HBV DNA; DS, double-strand HBV DNA replicative intermediates; SS, single strand HBV DNA replicative intermediates. C. Real-time PCR quantification of HBV pgRNA accumulation in post-transfection HepG2 cells. Results are expressed as the values normalized by GAPDH. D. Real-time PCR quantification of HBV cccDNA in post-transfection HepG2 cells. Results are expressed as the number of cccDNA copies per cell. E, F. The accumulation of HBsAg (E) and HBeAg (F) in post-transfection HepG2 culture supernatant was determined by ELISA, expressed as OD values. All results are expressed as mean ± standard from three independent analyses. *p < 0.05, **p < 0.01, ***p < 0.001 (One-way ANOVA).")
RXRα regulates HBV replication and transcription in HepG2. HepG2 cells were cotransfected with linear HBV genomic DNA and control pRS (HBV + Control vector), pRS-hRXRa (HBV + RXRα group), negative shRNA (HBV + Control shRNA) or RXRα shRNA vectors (HBV + RXRα shRNA), respectively. A. Real-time PCR quantification of cytoplasmic HBV DNA in post-transfection HepG2 cells. Results are expressed as the number of cytoplasmic DNA copies per cell. B. Southern blot analysis of cytoplasmic HBV replicative intermediates in HepG2 cells after 48 h transfection. The experiment was repeated thrice, and results of a representative assay shown. OC, open circular duplex HBV DNA; DS, double-strand HBV DNA replicative intermediates; SS, single strand HBV DNA replicative intermediates. C. Real-time PCR quantification of HBV pgRNA accumulation in post-transfection HepG2 cells. Results are expressed as the values normalized by GAPDH. D. Real-time PCR quantification of HBV cccDNA in post-transfection HepG2 cells. Results are expressed as the number of cccDNA copies per cell. E, F. The accumulation of HBsAg (E) and HBeAg (F) in post-transfection HepG2 culture supernatant was determined by ELISA, expressed as OD values. All results are expressed as mean ± standard from three independent analyses. *p < 0.05, **p < 0.01, ***p < 0.001 (One-way ANOVA).
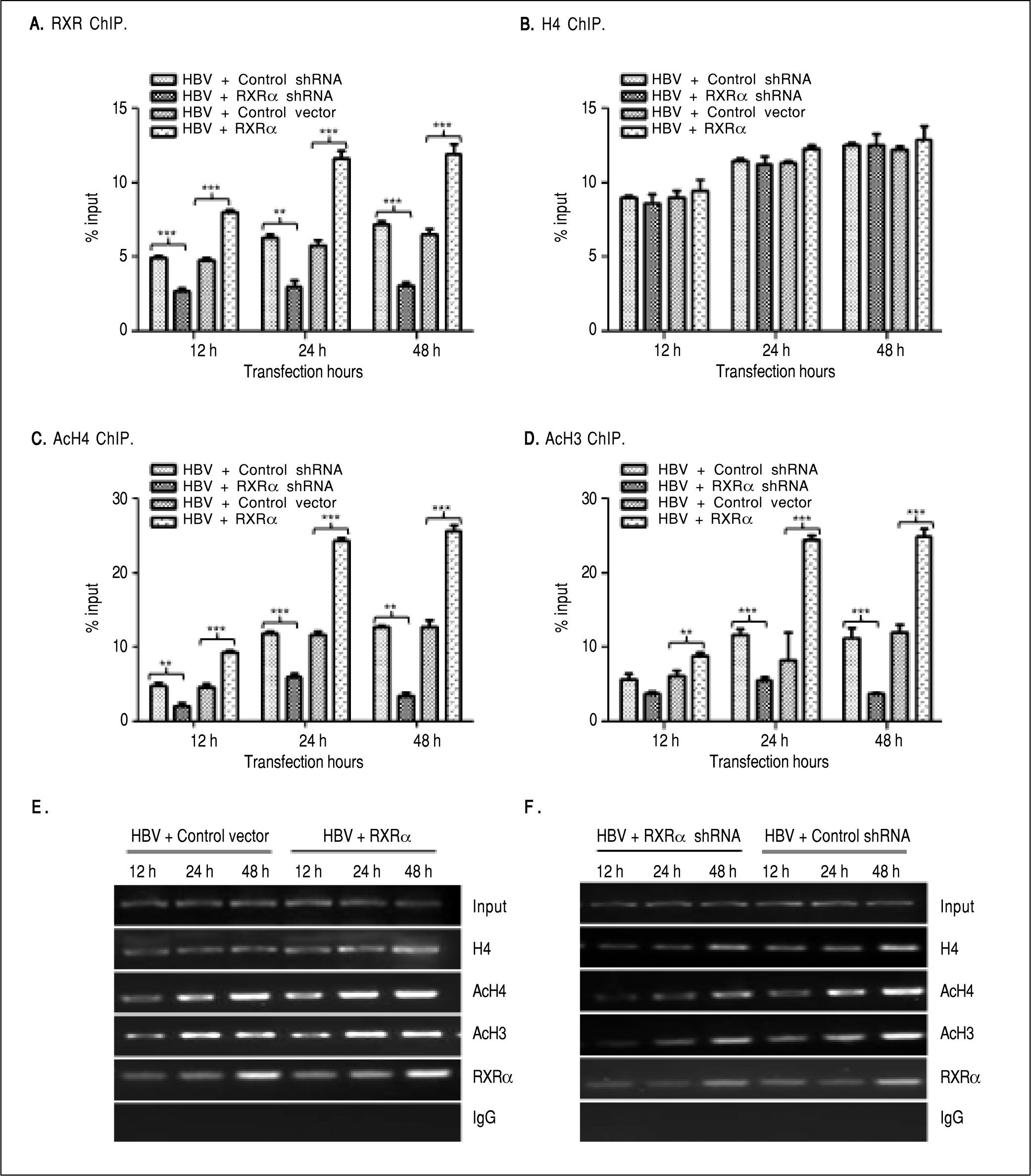
The binding of liver-enriched transcription factors to cis-elements of HBV DNA is a key step in the regulation HBV replication and transcription.10 To determine whether RXRα directly binds to HBV cccDNA, chromatin fragments of HepG2 cells transfected with linear HBV DNA were immunoprecipitated with the specific anti-RXRα antibody. As shown infigure 3 A and 3E, HBV cccDNA was successfully detected after pull down in the anti-RXRα chromatins. RXRa-HBV cccDNA binding was observed at 12 h and peaked at 48 h post-transfection, in parallel with HBV replication dynamics. RXRα overexpression or silencing significantly increased or decreased the recruitment of RXRα to HBV cccDNA in a time-dependent manner (Figures 3A and 3E), which demonstrated that RXRα directly bound to HBV cccDNA and subsequently regulated HBV replication.
, H4 (B), AcH4 (C), or AcH3 (D) antibody were analyzed by real-time PCR with primers and probes specific for HBV cccDNA. Each value was normalized to input DNA. The indicated numbers are % input with the ΔΔCT method. E and F. HBV cccDNA immunoprecipitated with RXRα, H4, AcH4 or AcH3 antibody was analyzed by endpoint PCR. The experiment was repeated thrice, and results of a representative as-say shown. * p < 0.05, ** p < 0.01, *** p < 0.001 (One-way ANOVA). H4: histone H4. AcH4: acetylhistone H4. AcH3: acetylhistone H3.")
RXRα regulates HBV minichromosome remodeling. The cotransfection of HepG2 cells are the same as indicated infigure 2. Chromatin DNA samples immunoprecipitated with RXRα (A), H4 (B), AcH4 (C), or AcH3 (D) antibody were analyzed by real-time PCR with primers and probes specific for HBV cccDNA. Each value was normalized to input DNA. The indicated numbers are % input with the ΔΔCT method. E and F. HBV cccDNA immunoprecipitated with RXRα, H4, AcH4 or AcH3 antibody was analyzed by endpoint PCR. The experiment was repeated thrice, and results of a representative as-say shown. * p < 0.05, ** p < 0.01, *** p < 0.001 (One-way ANOVA). H4: histone H4. AcH4: acetylhistone H4. AcH3: acetylhistone H3.
HBV cccDNA was organized into minichromosomes through interaction with histone and non-histone proteins and formed nucleoprotein complexes, whose state was closely related with viral replication and transcription.6,7,9 As shown infigures 3B and 3E, the binding of his-tone H4 to HBV cccDNA was detected at 12 h and peaked at 48 h after transfection with linear HBV DNA. Acetylated H4 and H3 histones in HBV minichromosomes were detected at 12 h and peaked at 48 h post-transfection, respectively (Figures 3C-3E). Our results are consistent with those from previous studies.8,9
To determine whether RXRα is involved in HBV minichromosome remodeling, HBV linear DNA was co-transfected with RXRα expression or shRNA vectors into HepG2, and levels of recruitment and acetylation of his-tones in the viral minichromosome were quantified. As shown inFigures 3B, 3E, and 3F, RXRα overexpressionor silencing did not affect the recruitment of histone H4 to cccDNA. However, RXRα overexpression significantly increased the acetylation of histones H4 and H3 in the viral minichromosome (Figures 3C, 3D, and 3E). When RXRα was knocked down in HepG2, the acetylation of histones H4 and H3 was dramatically (p < 0.01) suppressed, as indicated infigures 3C, 3D, and 3F. These results suggest RXRα involvement in the regulation of HBV minichromosome remodeling.
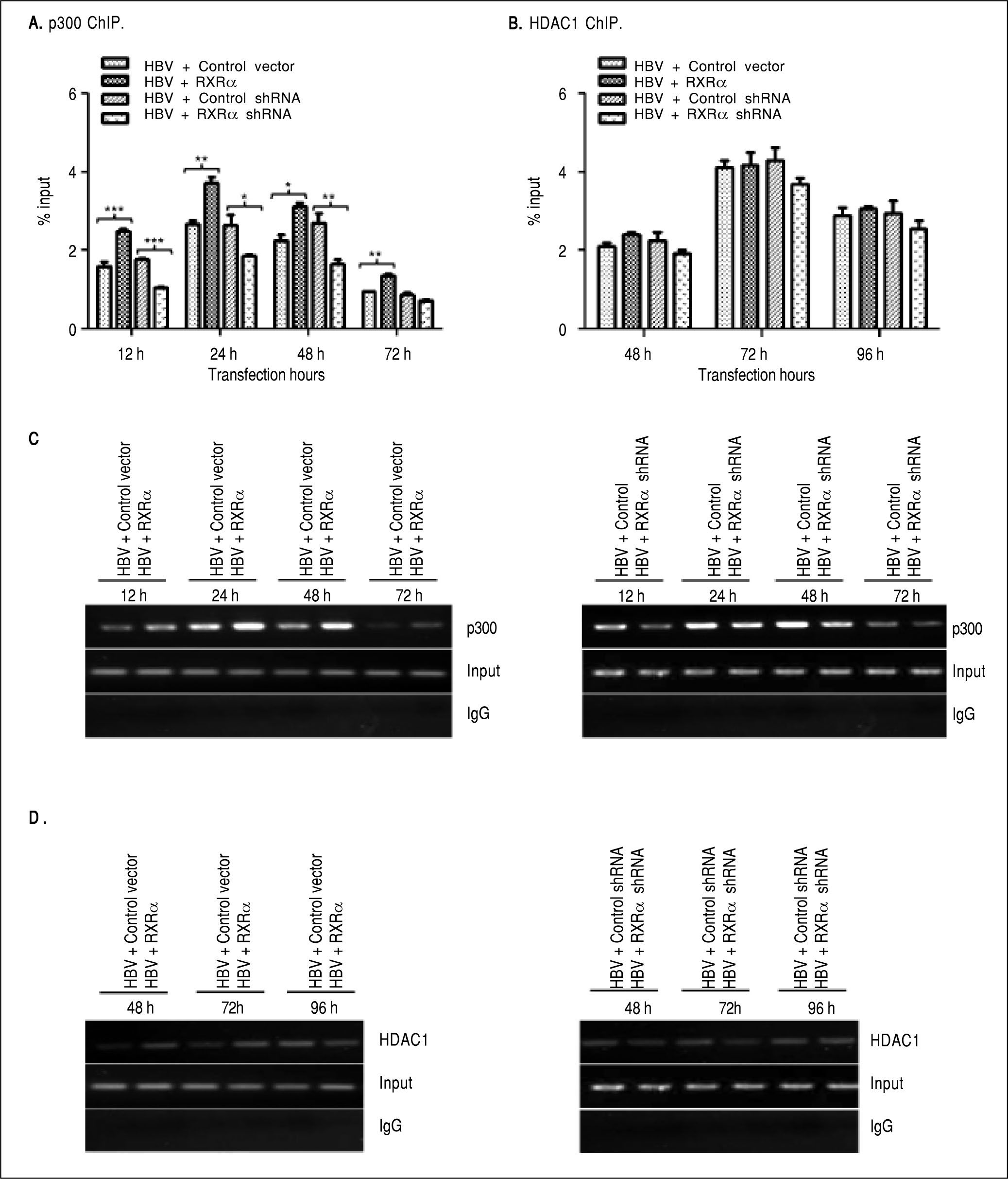
RXRα modulates HBV minichromosome remodeling by affecting the recruitment of chromatin-modifying enzymesPrevious studies observed that histone acetyltransferase and deacetylases were recruited onto HBV cccDNA in a different time-dependent manner during HBV replication.7,9 To assess whether RXRα contributes to the recruitment of chromatin-modifying enzymes, chromatin fragments of HepG2 co-transfected with linear HBV DNA and RXRα expression or shRNA plasmids were immunoprecipitated with p300 or theHDAC1 antibody. As shown infigure 4A and 4C, the recruitment of p300 onto cccDNA was observed at 12 h, peaked at 24 h, and declined thereafter. p300 recruitment was strongly elevated during RXRα overexpression and decreased during silencing of RXRα, which closely paralleled the kinetics of histone acetylation and HBV replication. Previous studies have shown very low HDAC1 recruitment at 48 h post-transfection;7,9 therefore, we investigated the state of cccDNA-bound HDAC1 in HepG2 between 48 and 96 h after co-transfection. As indicated infigures 4B and 4D, while the recruitment of HDAC1 onto cccDNA was detected at 48 h and significantly increased at 96 h after transfection of HBV linear DNA, RXRα overexpression and knock-down showed no significant influence on HDAC1 binding with cccDNA. Taken together, these results indicated that RXRα is involved in the recruitment of p300 but not ofHDAC1 onto cccDNA during HBV replication.
.")
The effects of RXRα on the recruitment of p300 and HDAC1 onto HBV cccDNA HepG2 cells were cotransfected vectors as indicated infigure 2. A, B. Real-time PCR quantification of HBV cccDNA immunoprecipitated by p300 and HDAC1 antibody. All data expressions and normalizations are the same as indicated infigure 3. C, D. Endpoint PCR quantification of HBV cccDNA immunoprecipitated by p300 and HDAC1 antibody. The experiment was repeated thrice, and results of a representative assay shown. *p < 0.05, **p < 0.01, ***p < 0.001 (One-way ANOVA).
RXRα behaves as a miscellaneous nuclear receptor that activates transcription of target genes in different metabolic pathways.19 Approximately 16% of hepatic expressed genes are shown to be regulated in an RXRα-dependent manner.20 The HBV genome contains four open reading frames (coding for surface, core, polymerase, and X proteins), four promoters (pre-S1, pre-S2, core, and X) and two enhancers (enhancer 1 and enhancer 2).10,21,22 Previous studies have indicated that RXRa directly binds to the HBV enhancer 1 and core promoter and modulates HBV transcription and gene expression.13,16,23 RXRa appears to function through forming a homo- or a heterodimeric complex with other nuclear receptors.19,24 The retinoic acid receptor α (RARα)-RXRα heterodimer has been shown to modulate chromatin remodeling of target genes through binding to RA response elements (RAREs).25,26 The activity of the RAR-RXR heterodimer is influenced by its physiological ligands, such as retinoic acid (RA). The heterodimer recruits the HDAC in the absence of RA, but associates with transcriptional coactivators including proteins possessing histone acetyltransferase (HAT) upon RA addition, suggesting that it regulates the transcription of target genes by altering chromatin structure.19,25-27
In this study, coupling cccDNA real-time PCR and cccDNA ChIP assay, we found that RXRα was recruited onto the HBV minichromosome. The RXRα recruitment on the cccDNA was paralleled with HBV replication as well as histone recruitment and acetylation in the cccDNA minichromosome. We also found that both p300 and HDAC1 were recruited onto HBV cccDNA, which is consistent with results from previous work. Although RXRα overexpression or silencing significantly increased or impaired the recruitment of the p300 acetyltransferase, it showed no significant influence on the recruitment of HDAC1. Our results imply that RXRα activates HBV replication by directly binding to the cccDNA and trig-geringcccDNA epigenetic changes by modulating the recruitment of acetyltransferase onto the viral minichromosome. Although we demonstrated the interactions and effects among RXRα, p300, and HBV cccDNA, the mechanism epigenetics and the recruitment of chromatin-modifying enzymes remain unclear. Further studies are needed to clearly illustrate the underlying details.
The persistence of HBV cccDNA in the nuclei of infected cells ensures a stable resource of pgRNA for replication and provides templates for mRNA synthesis and viral protein production.2,4 HBV cccDNA has been proven relatively insensitive to currently available antiviral drugs and is thought to be a key part of persistent HBV infection and treatment failure.528-30 Therefore, there is a need to identify novel antiviral drugs acting on the cccDNA as an alternative treatment choice. The factors (e.g., RXRα) involved in regulating the HBV cccDNA minichromosome may be potential targets in such drug development.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Grant No. 81071338).
DisclosuresThe authors declare that they have no competing financial interests.



