



The liver has a remarkable ability to regenerate in response to surgical removal or chemical insult. The mechanisms regulating regenerative processes are complex, and incompletely understood. A large number genes, which are not normally expressed in the quiescent liver, are activated. Immediately after partial hepatectomy (PH) (1-6 h), nitric oxide (NO) is synthesized by liver parenchymal and nonparenchymal cells from L-arginine, via induction of the inducible form of nitric oxide synthase (iNOS). NO is a highly reactive molecule, known to be involved in diverse biological processes in nearly all aspects of life. Liver regeneration is a major area within the field of NO research. Our review describes several processes that have been suggested to be modulated by the NO released following PH, including proliferation, apoptosis and angiogenesis in the remnant tissue. Because iNOS up regulation has such profound physiologic effects, its regulation is strictly controlled. The up regulation of iNOS after PH and the subsequent production of NO induce positive effects on the regulation of early stages of the regenerative process. However, overproduction (> 100%) can have detrimental effects, including apoptosis. Thus, the iNOS induction after PH is necessary, and enough to allow for the normal regenerative process.
Liver regeneration after the loss of hepatic tissue is critical for the restoration of the homeostatic role of the organ. Loss of liver mass can be induced by administering hepatotoxic chemicals (e.g. carbon tetrachloride). This is followed by an inflammatory response, which removes tissue debris, followed by the regenerative response. Most commonly, however, regeneration of the liver is studied by performing a surgical procedure which removes 2/3 of the liver mass in rodents (rats and mice), a technique known as 2/3 partial hepatectomy (PH).1,2 Due to the multi-lobe structure of the rodent liver, three of the five liver lobes (representing 2/3 of the liver mass) can be removed by an easy surgical procedure, without causing any tissue damage to the residual two lobes. The latter grow in size to restore an aggregate equivalent to the mass of the original five lobes. The process, in rats and mice, is complete within 5-7 days after surgery.3 The reproducibility of PH in terms of mass removed and precision of timing of the sequence of ensuing events has made PH the preferred approach for experimental study of liver regeneration. In a clinical setting, this procedure is also done in humans, in order to resect solitary liver metastases or repair trauma, etc.4
After PH exist a rapid and synchronized compensatory regenerative response in the remaining tissue that increases cell number and reinstates organ function. PH triggers a sequence of events that proceed in an orderly fashion and can be observed from the first 5 min to 5-7 days. Hepatocytes are the first cells to enter into DNA synthesis. A 2/3 PH leaves a residual 1/3 of hepatocytes. They undergo one round of DNA synthesis (leading to 60% of hepatocytes) which peaks at 24 h for the rat and at approximately 36 h for the mouse. A second smaller percent of cells enter into a second round of DNA synthesis and establish the original number of hepatocytes. A small wave of apoptosis of hepatocytes seen at the end of DNA synthesis suggests that this is a mechanism to correct an over-shooting of the regenerative response.3
One remarkable characteristic of the regenerative process after PH is the capacity of the liver to grow until the organ size and functionality are fully restored. Therefore, it is widely accepted the existence of a precise pattern of events (release and modulation of growth factors and cytokines) controlling the successive regenerative steps after PHx.1
PH induces rapid induction of more than 100 genes not expressed in normal liver.5 These genes relate directly or indirectly to preparative events for the entry of hepatocytes into the cell cycle. The precise role of the many genes expressed early in liver regeneration is not always clear and the early changes in gene expression should be viewed as serving both the entry of hepatocytes into the cell cycle as well the orchestration of specific adjustments that hepatocytes have to make, so that they can carry all essential hepatic functions while going through cell proliferation.1,4,5
The events occurring in the early period of 0-1 h after PH have often been called “priming”.1 The term is a useful one, in that it denotes not only events associated for preparation for entry into the cell cycle, but also events and strategies of hepatocytes aimed at modifying patterns of gene expression so that they continue to deliver their homeostatic functions. During this phase, the initial factors comprise interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-a). Following IL-6 binding to the gp130 receptor, activation of STAT 3 and C/EBP beta/nuclear factor-IL-6 takes place. Both cytokines TNF-a and IL-6 triggers the transition G0/G1 in the cell cycle. IL-6 and TNFR1 deficient animals fail to accomplish initiation and regenerative response.1,6,7 Another change in the immediate hours following PH is the in vivo induction of nitric oxide synthase and the release of nitric oxide (NO).2,8
Following to priming/initiation, several immediate early-phase genes related to hepatocyte proliferation are induced within 2 h. They comprise c-fos, c-jun and others. The progression of the priming/ competent hepatocytes through G1 and subsequent replication depends on the signaling mediated by hepatocyte growth factor (HGF), transforming growth factor alpha (TGFa) and epidermal growth factor (EGF) by another.4,7,9,10 Then, progression in the cell cycle is regulated by the expression of cyclins and cyclin-dependent kinase.11
Nitric OxideNitric oxide moleculeNitric oxide (NO), a short-lived, highly reactive free radical, influences physiological processes in virtually every organ and tissue. It exhibits a remarkably broad spectrum of functions, including neurotransmission and memory formation, regulation of blood pressure, mediation of the bactericidal and tumoricidal activity of macrophages, and liver regeneration.
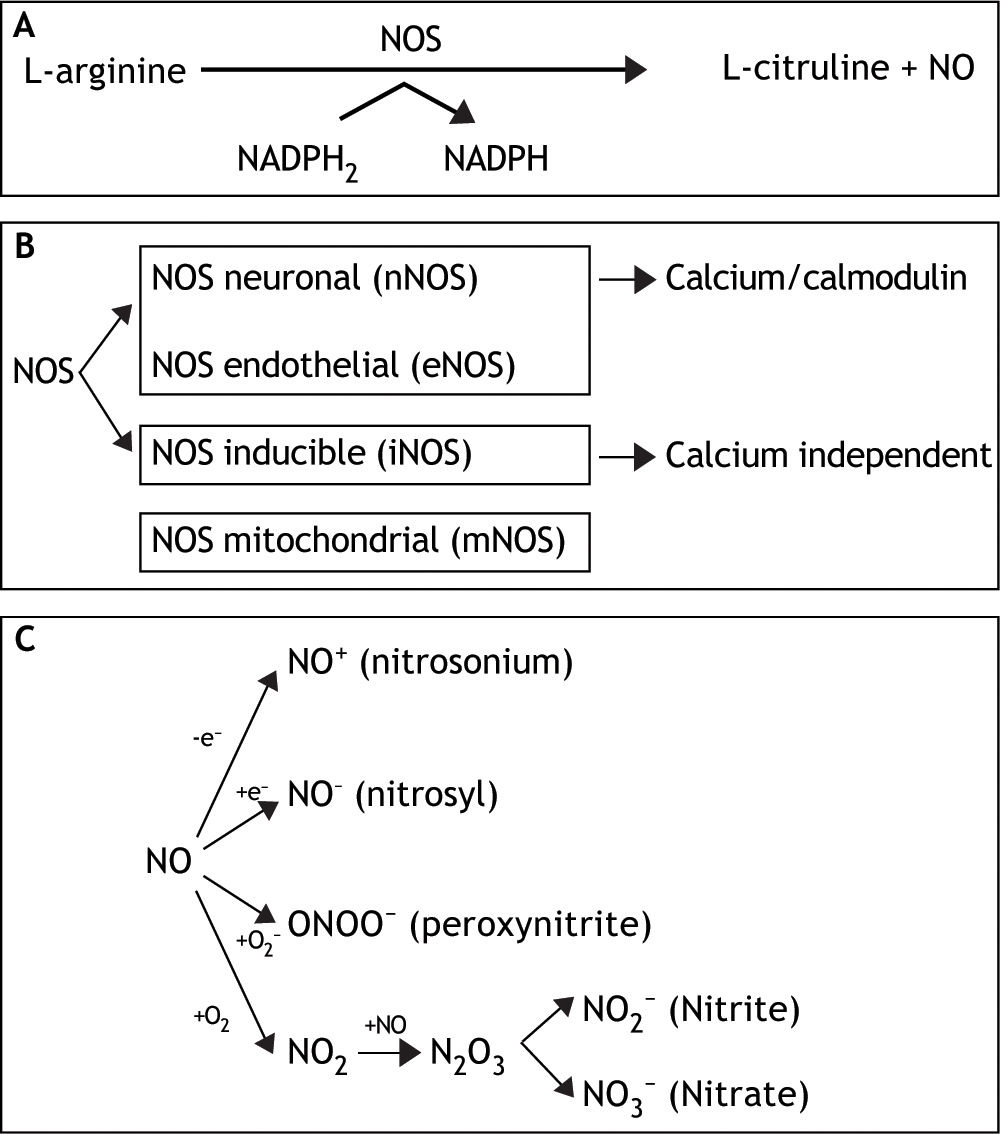
This small, unstable, gaseous, free radical performs its complex tasks by acting as an intra-or extracellular messenger molecule. Nitric oxide is produced from the amino acid L-arginine through a reaction catalysed by the enzymes nitric oxide synthases (NOS). These enzymes catalyse the oxidation by molecular oxygen of one of the guanidine nitrogens in L-arginine to form NO and citrulline12 (Figure 1A).
. Distinct isoforms of NOS are presented. C. Under normal conditions, interaction of NO with oxygen results in the formation of the relatively inactive end products nitrite (NO2-) and nitrate (NO3-). NO can be also converted into different reactive chemical forms as nitrosonium (NO+) and nitroxyl (NO-) radicals, by gaining or losing electrons, respectively. Finally, NO can combine with O2 - (superoxide anion) to render ONOO- (peroxynitrite).")
A. NO is synthesized endogenously by conversion of L-arginine to L-citruline, using NADPH as an electron donor. B. The reaction is catalyzed by a family of enzymes called nitric oxide synthases (NOS). Distinct isoforms of NOS are presented. C. Under normal conditions, interaction of NO with oxygen results in the formation of the relatively inactive end products nitrite (NO2-) and nitrate (NO3-). NO can be also converted into different reactive chemical forms as nitrosonium (NO+) and nitroxyl (NO-) radicals, by gaining or losing electrons, respectively. Finally, NO can combine with O2 - (superoxide anion) to render ONOO- (peroxynitrite).
- •
NO synthase isoforms and subcellular localization. Three main NOS isoforms have been identified and cloned so far: nNOS (neuronal or type I), iNOS (inducible or type II), and eNOS (endothelial or type III). In addition, new isoforms or mitochondrial variants of NOS (mtNOS) have been described recently in rat liver, thymus, and brain.13 eNOS and nNOS are constitutively expressed, and produce relatively small amounts of NO. These isoforms depend on Ca2+/calmodulin, and account for a rapid increase of NO in response to hormone receptor interaction.14,15 On the other hand, iNOS is up-regulated in liver under a number of conditions, including endotoxemia, hemorrhagic shock, ischemia-reperfusion, hepatitis and liver regeneration. This isoform is Ca2+-independent, and synthesizes NO for extended periods of time, at high concentrations, serving as an important regulator and effector during inflammation and infection.16 It is expressed in different types of cells, including endothelial cells, hepatocytes, Kupffer cells and smooth-muscle cells.
NOS isoforms have differential localizations within the cells: nNOS and iNOS are mainly cytosolic, although recently, Stolz, et al. have established the peroxisome as a site of iNOS localization in hepatocytes.17 On the other hand, eNOS has been identified at the plasma membrane, rough endoplasmic reticulum and in the nuclei of rat hepatocytes, and has been shown to be a membrane associated protein.18 Finally, mtNOS activity is modulated by Ca2+, and it is located at the inner mitochondrial membrane, where continuously controls mitochondrial respiration.19 In liver, eNOS activity is normally detectable in Kupffer cells and at the plasma membrane of rat hepatocytes.7 In addition, functional eNOS has been identified and characterized in sinusoidal endothelial cells with the liver and may contribute to local distribution of perfusion and portal pressure.20
In addition, hepatocytes, Kupffer and stellate cells are prompted to express an intense iNOS activity once exposed to effective stimuli, such as bacterial lipopolysaccharide (LPS) and cytokines1 (Figure 1B).
- •
NO and biological reaction. Reactive nitrogen intermediates (RNI) are now also recognized as important radicals. Under normal conditions, interaction of NO with oxygen results in the formation of the relatively inactive end products nitrite (NO2-) and nitrate (NO3-). NO can be also converted into different reactive chemical forms (NO-, NO. and NO+), and this is the reason why it has a wide range of chemical reactivity and regulatory functions in a variety of biological targets (Figure 1C). These functions include regulation of the cardiovascular system, smooth muscle relaxation, neurotransmission, coagulation and immune regulation. Despite these beneficial functions, the molecule has also a pivotal role in cell death, by having the ability to either induce or protect against apoptosis depending on its levels and cellular context. Furthermore, NO can turn an apoptotic response into a necrotic one. NO can also combine with O2- (superoxide anion) to form peroxynitrite (ONOO-), which shares some properties with NO, such as its capability to freely diffuse via the intra-and the intercellular pathway, and also to act as a powerful oxidant.21
Apoptosis, or programmed cell death, is essential to the normal development of multicellular organisms as well as to the physiologic cell turnover. NO is one of the most potent regulators of apoptosis since, as stated above, it has dual pro-and anti-apoptotic effects. The cellular threshold for apoptosis is highly regulated, especially by members of the Bcl-2 protein family. Members of this family are anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-w) while others can promote programmed cell death (Bax, Bak, Bad, BNIP3).22 The oligomerization of pro-apoptotic proteins into the outer mitochondrial membrane leads to the formation of pores, and results in mitochondrial release of cytocrhome c. The association of cytochrome c with an adapter molecule, Apaf-1, and caspase 9 in the cytoplasm activates the latter, which, in turn, activates downstream caspases.23
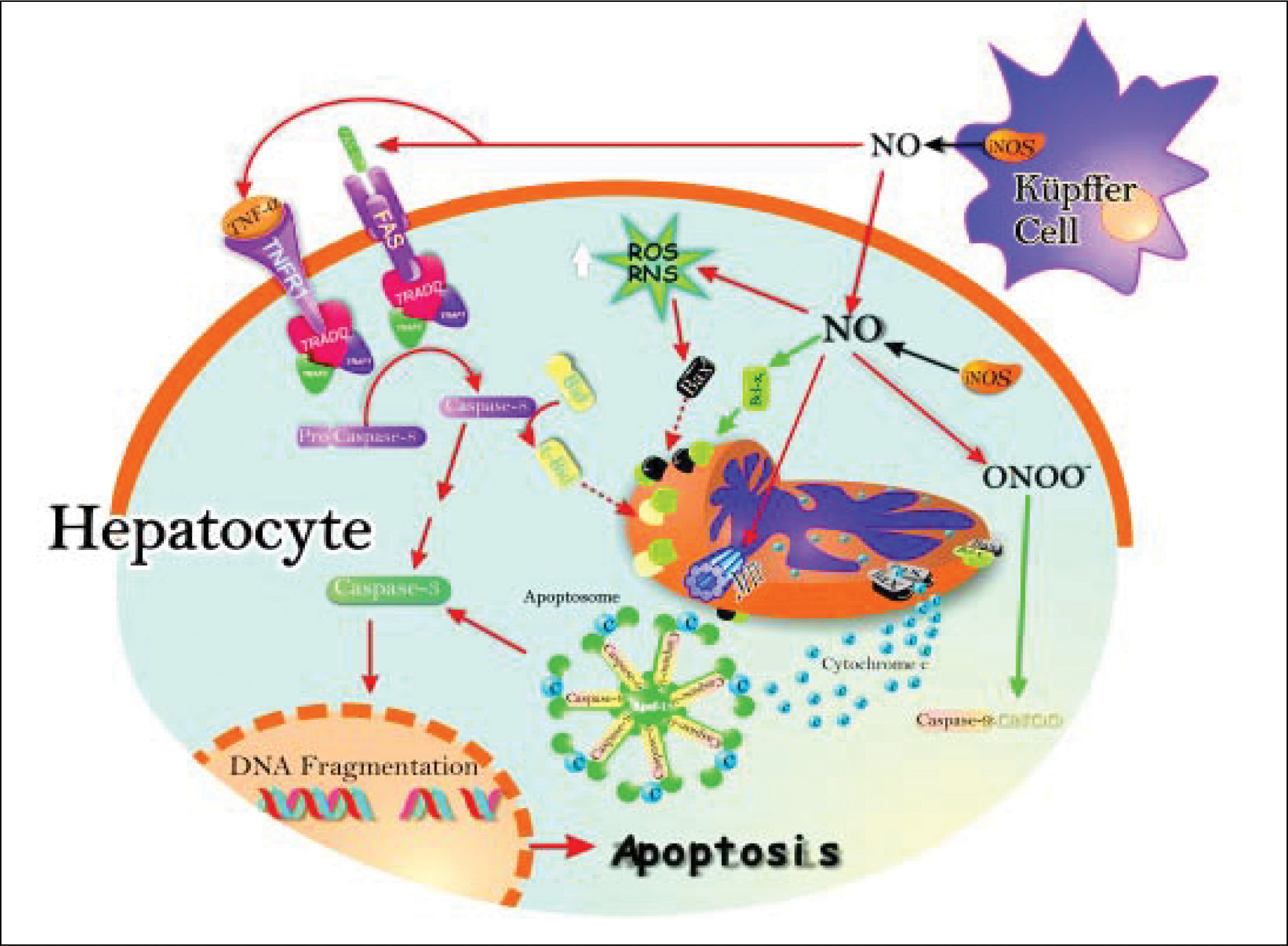
In addition, apoptosis can be induces by the TNF-induced death receptor pathway. They have been identified two families of receptor: the receptor of TNFa (TNFR1) and Fas receptor protein that induce apoptosis in a similar way. Binding of TNFa to TNFR1 results in receptor trimerization and the recruitment of a series of intracellular proteins24 that ultimate bind caspase-8, leading to its activation.25 Activated caspase-8 initiates a proteolytic cascade that results in release of lysosomal cathepsin B, cleavage of the pro-apoptotic Bcl-2 family member Bid, initiation of the mitochondrial death pathway with release of cytocrome c, and activation of downstream effectors caspases that ultimately induces apoptosis26 (Figure 2).
 which may raise pro-apoptotic protein Bax levels, and downstream, cytochrome c release and activation of the apoptosome. Peroxynitrite can open membrane transition pores (MTP), and lead to cytochrome c release (red arrows). The inhibition of apoptosis by NO is the result of S-nitrosylation of active caspase 9, which attenuates caspase activity and thereby blocks substrate cleavage. Also, the induction of anti-apoptotic protein Bcl-xL by NO leads to inhibition of cytochrome c release to cytosol, which in turn inhibits apoptosis (black arrows).")
Schematic representation of the pro-and antiapoptotic effects exerted by NO. The NO production by up-regulation of iNOS in both hepatocytes and Kupffer cells leads to Fas stimulation. This produces caspase 8 activation, followed by increase of caspase 3 activity and, downstream, release of cytochrome c from mitochondria to cytosol, with further activation of the apoptosome. Caspase 8 activation may promote Bid processing, leading eventually to apoptosis. Once peroxynitrite is increased, it produces increase of reactive nitrogen intermediaries (RNI) which may raise pro-apoptotic protein Bax levels, and downstream, cytochrome c release and activation of the apoptosome. Peroxynitrite can open membrane transition pores (MTP), and lead to cytochrome c release (red arrows).
The inhibition of apoptosis by NO is the result of S-nitrosylation of active caspase 9, which attenuates caspase activity and thereby blocks substrate cleavage. Also, the induction of anti-apoptotic protein Bcl-xL by NO leads to inhibition of cytochrome c release to cytosol, which in turn inhibits apoptosis (black arrows).
NO induces apoptosis both in vivo27,28 and in several cell types in vitro, such as neuronal cells,23 macrophages,29 cardiac myocytes,30 endothelial cells,31 lymphocytes, and thymocytes.32 The mechanisms of NO-induced apoptosis are presently under intensive investigation and several mechanisms underlying the effects of NO on apoptosis have now been elucidated. They include activation of the death receptor Fas through upregulation of Fas ligand expression, generation of the potent oxidant and cytotoxic mediator peroxynitrite, inhibition of mitochondrial ATP synthesis, and inactivation of several antioxidant enzymes.33,34
Another site of action of NO on mitochondria is the mitochondrial permeability transition pore (MPTP). There is now increasing evidence that support a redox regulation of cytochrome c release during apoptosis.35 The MPTP plays an essential role in apoptosis; once opened, cytochrome c is released, resulting in the formation of the apoptosome, activation of caspase-9 and execution of apoptosis. Recently, inhibition of MPTP opening was identified as a novel site of action for NO signaling in apoptosis.36 Inhibition of MPTP opening would result in less cytochrome c available to initiate apoptosis. This study suggests that a fine balance exists between the pro-and anti-apoptotic properties of NO at the level of the mitochondrion.
Apart from pro-apoptotic action, NO may protect apotosis death under certain conditions, depending on the cell type.
Several mechanisms have been proposed to elucidate the ability of NO to confer protection against apoptotic cell death. These can be divided into cGMP-dependent and cGMP-independent mechanisms:
- •
cGMP dependent mechanisms. It is known that NO mediates many of its physiological functions through the direct heme-dependent activation of soluble guanilate cyclase, and the consequent increase in intracellular cGMP levels.37 NO-dependent generation of intracellular cGMP has been shown to protect against apoptosis in lymphocyte,38 eosinophils,39 embryonic motor neurons.40 Similarly, an increase of cGMP suppresses apoptosis in hepatocytes through a marked activation of serine/threonine kinase Akt, suggesting a link between cGMP and the PI3K/Akt signaling pathway.37,41 Activation of Akt can promote cell survival through phos-phorylation of the pro-apoptotic protein Bad and caspase 9, which favors Bad proteosomal degradation of Bad and caspase 9 inactivation, respectively, and by preventing cytochrome c release from mitochondria.42,43
Among the other genes regulated by NO is BNIP3, a protein known to promote apoptosis, which belongs to the Bcl-2 family. BNIP3 localizes at the mitochondria and other cytoplasmic membrane structures, and is found widely expressed several mouse and human tissues. BNIP3 expression is markedly suppressed following iNOS up regulation and NO production in a cGMP-dependent manner, suggesting that this is another mechanism by which NO prevents apoptosis mediated by cGMP-dependent pathways.44 Ray, et al. demonstrated recently that BNIP3 heterodimerizes with Bcl-2/Bcl-xl and induces cell death independent of a BH3 domain at both mitochondrial and non-mitochondrial sites.45
- •
cGMP-independent mechanisms. NO protects apoptotic cell death in a cGMP-independent manner by preventing Bcl-2 cleavage, cytochrome c release, and the induction of protective proteins such as Hsp70 and Bcl-2.41 In addition, many of the direct protective actions of NO are mediated by S-nitrosylation of proteins. S-nitrosylation involves the transfer of a nitric group to cysteine sulfhydryls, leading to the formation of a nitrosothiol (RSNO). While the activity of NO is often limited due to its very short half-life, nitrosothiols can be very stable compounds. Caspases contain a highly conserved cysteine residue within their active site, and therefore are a target for inactivation via S-nitrosylation. In line with this, NO has been reported to protect against Fas-induced liver injury by inhibiting caspase activity. This caspase inhibition is reverted by DTT, suggesting that cysteine S-nitrosylation is the underlying mechanism of caspase regulation by NO in this study.46
Under normal conditions, only the constitutive eNOS is present in the liver, and the low level of NO produced by eNOS regulates hepatic perfusion, among other functions. However, iNOS is readily up-regulated in the liver under a number of conditions, including endotoxemia, hemorrhagic shock, ischemia-reperfusion, sepsis, infection, hepatitis, ozone exposure, and liver regeneration.47 Once iNOS is expressed, large amounts of NO are generated in the liver in a sustained fashion, which functions as an important regulator and effector during inflammation and infection.48 Because the liver plays a crucial role in m any metabolic and immune processes, physiological and pathophysiological functions of the NO generated in the liver have prompted numerous investigations in recent years. Both cytoprotective and cytotoxic effects of NO have been demonstrated in the liver.37,48,49
- •
Induction of hepatic iNOS after different stimulus. In response to endotoxin or proinflammatory cytokines, such as tumor necrosis factor-a (TNF-a), interleukin (IL)-1 and interferon-Y, as well as their combinations, iNOS is rapidly up-regulated within hours in hepatocytes, and in resident hepatic macrophages (Kupffer cells) as well.50 These stimuli often act synergistically to induce iNOS expression; however, IL-1ß alone is an effective stimulator of iNOS in liver. The cytokine-mediated up-regulation of iNOS gene transcription requires the transcriptional factor nuclear factor-KB (NF-KB) in both animals and humans.51 In addition, hepatic endothelial cells and stellate cells can also produce NO through iNOS up regulation. Therefore, in liver inflammation, hepatocytes are situated in an environment where NO is generated from surrounding cells, as well as from hepatocytes themselves. In cell cultures has been reported that the maximum production of NO, which is calculated as the release of nitrite, after stimulation with polysaccharides and/or cytokines in hepatocytes is between 45 and 1,200 nmol NO2-/106 cells per day, Kupffer cells between 50 and 100 nmol NO2-/106cells per day, and endothelial cells between 10 and 16 nmol per day NO2-/106cells37,52 (Figure 2).
- •
NO and liver regeneration. One remarkable characteristic of the regenerative process after PH is the capacity of the liver to grow until the organ size and functionality are fully restored. Therefore, it is widely accepted the existence of a precise pattern of events (release and modulation of growth factors and cytokines) controlling the successive regenerative steps after PH.1 However, the nature of the factors and early signals involved in the recruitment of cells to entry in the division cell cycle is far from been fully understood. The current view is based on the existence of a dynamic balance between positive and negative control. Equilibrium between stimulator and inhibitor gene of the cell cycle expressed after PH, may explain why liver regeneration is tightly regulated growth process. Within 30 min after PH, several genes are induced which contributes to regeneration. Among these genes, signal transducer and activation of transcription-3 (Stat-3), nuclear factor kB (NF-kB), CCAAT/enhancer binding protein b (C/ EBPb), and activating protein 1 (AP-1) are known to play a cooperative role in intracellular signaling cascades leading to DNA synthesis.53-55 These transcription factors regulate the expression of many hepatocyte genes including iNOS,53,56 which is up-regulated within the immediate hours after PH. NO begins to be released within 30 min after PH, reaching its maximal level at 5 h (progression phase of cell cycle) and returns to basal levels at 18 h after surgery.1,33 In this connection, our group has demonstrated a marked decrease in the peak of DNA synthesis in hepatectomised rats when they were pretreated with two iNOS inhibitors (a specific inhibitor, aminoguanidine, and a non-specific one, NG -mo-nomethyl-L-arginine, L-NAME).8 Likewise, Rai, et al. have shown impaired liver regeneration in iNOS-deficient mice.57 These results suggest a positive effect of NO in the regulation of the regenerative process at early stages.
Interestingly, this NO seems to be delivered exclusively in the liver, and apparently, the molecule is completely consumed in the hepatic tissue. This conclusion is supported by the observation of a total absence of NO in blood, as measured through the formation of a nitroxyl-hemoglobin complex, as well as by the absence of changes in the nitrite concentration in plasma, a NO-derived metabolite that is more stable than NO itself.2 After PH, both hepatic iNOS activity and levels of iNOS messenger RNA have been detected exclusively in the liver, indicating that this is a local effect.2,8,12 The relative contribution of each liver cell type (Kupffer cells, and hepatocytes and, possibly, endothelial cells) to the total iNOS activity and NO synthesis seems to be different. However, because NO can easily diffuse through the cells, the origin of this molecule is not critical for the ability to promote intracellular changes in neighboring cells.2
Evidence shows the importance of cytokines during liver regeneration. When a large piece of liver is removed by PH, increased local expression of TNF-a triggers the production of another cytokine, IL-6, and both cytokines are required to initiate subsequent hepatocyte proliferation.58In vivo analysis of murine iNOS genes indicate that neither TNF-a nor IL-6 alone are sufficient to activate iNOS transcription, but when these two cytokines are combined, up-regulation occurs.59,60
- ø
Role of NO in apoptosis/proliferation balance. The determinants of hepatocytes proliferation during liver regeneration are highly complex, and different mechanisms operate between initiation of DNA synthesis and the termination of proliferative process.61 The control of liver regeneration and the events involved in regulating the growth of the organ remain unknown. It has become increasingly apparent that apoptosis plays a key role in the cell cycle.62,63 Many of the proteins that can induce cell death are components of the cell division cycle.41 In previous studies by our group, we analyzed the role of NO in the regulation of apoptosis/survival-related proteins.48,63
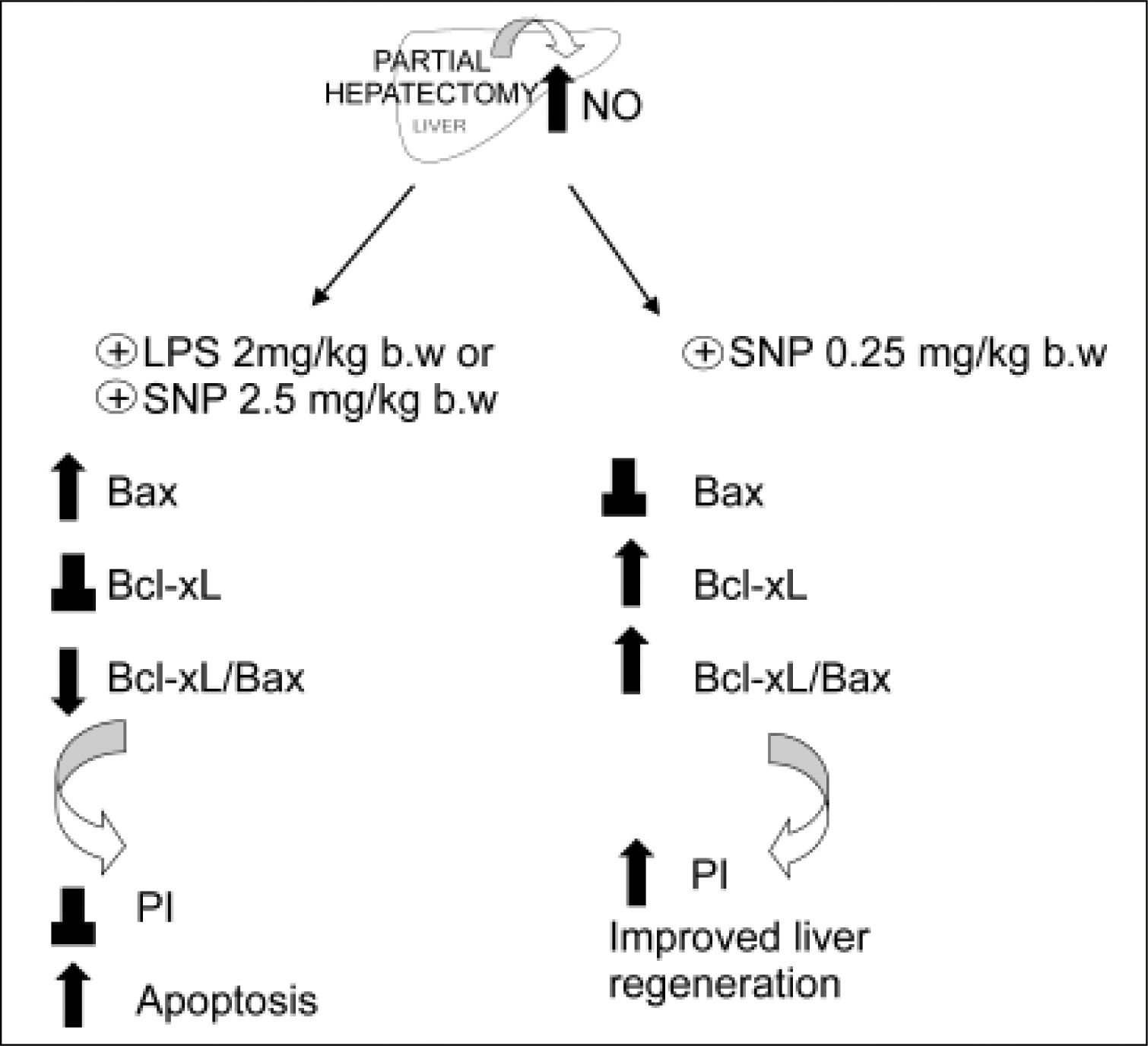
The relative prevalence of Bax and Bcl-xL proteins are critical factors influencing cell fate; they promote either cell survival or death, whose ultimate outcome largely depends on the Bcl-xL/Bax ratio.63 Large increases of the hepatic levels of NO (> 100%) (obtained either by the iNOS inducer LPS or by the direct NO donor sodium nitroprusiate (SNP, 2.5 mg/kg body wt, administered 5 h after PH) induces expression of the pro-apopoptotic protein Bax in the mitochondrial fraction, without changes of the anti-apoptotic protein Bcl-xL as compared with PH-alone.48 On the contrary, when the increase of NO is moderate, e.g. a 35%, obtained by low doses of SNP (0.25 mg/kg body wt), the pro-apoptotic protein Bax remains unchanged with respect to PH-alone, while the anti-apoptotic protein Bcl-xL is increased (Ronco, et al., unpublished data).
The mechanism by which NO produces changes of Bax/Bcl-xL gene expression remain unknown. Some investigators have focused on p53 as a linkage between NO and Bax/Bcl-2 genes, since NO is known to induce p53 accumulation, which is a direct transcriptional activator of Bax gene and a transcriptional inhibitor of Bcl-2 gene.64 p53 acts as a checkpoint control of the cell cycle, permitting the repair of damaged DNA. The blockage in G1/S transition that results from p53 activation has been suggested to cause apoptosis in the case of severe DNA damage. Expression of wild-type p53, a tumor suppressor gene, seems to be closely linked with apoptosis caused by most of DNA-damaging agents.65,66 Recent data from our laboratory showed that a large augmentation of NO in the liver after PH (as those caused by LPS and SNP 2.5 mg/ kg body wt.), increases p53 protein levels with a correlation between cytosolic nitrate and p53 levels.48 On the other hand, a smaller increase of NO (as that caused by SNP 0.25 mg/kg body wt) does not show differences in p53 expression when compared with hepatectomized rats (Ronco, et al., unpublished data) (Figure 4). The apoptotic index revealed that a high increase of NO induces cellular death by apoptosis, while a mild increase does not modify this process. In agreement with other authors,64,65 our results suggest that high levels of NO induce apoptosis by accumulation of p53, which induces Bax expression.48
 and the treatment with NO donor SNP2 (2.50 mg/mL) produce an over-increase of 100% of NO, leading to an increase of pro-apoptotic proteins Bax and p53, leading to apoptosis. Treatment with a NO donor, SNP1 (0.25 mg/mL), produces an increase lower than 35% of NO, thus leading to an increase of anti-apoptotic protein BclxL, which improves liver regeneration (⊥ not difference, ↑ increase, ↓ decrease).") Figure 3.
Figure 3.Schematic representation of the effects of the overproduction of NO after PH. The induction of iNOS with lipopolisacaride (LPS, 2 mg/kg) and the treatment with NO donor SNP2 (2.50 mg/mL) produce an over-increase of 100% of NO, leading to an increase of pro-apoptotic proteins Bax and p53, leading to apoptosis. Treatment with a NO donor, SNP1 (0.25 mg/mL), produces an increase lower than 35% of NO, thus leading to an increase of anti-apoptotic protein BclxL, which improves liver regeneration (⊥ not difference, ↑ increase, ↓ decrease).
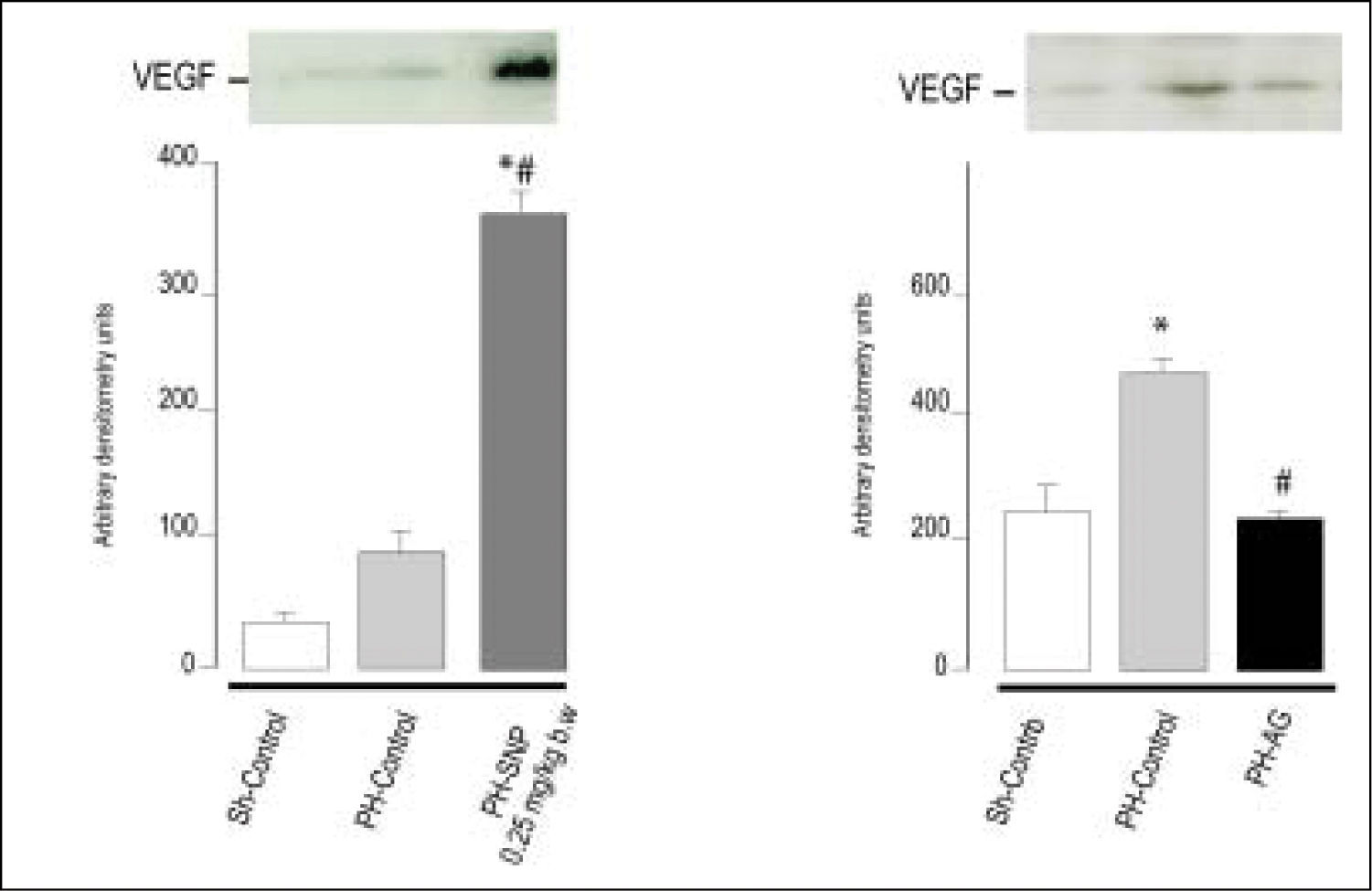
(0.11MB). 100 mg/kg body weight, intraperitoneally. *p < 0.05 vs. Sh-Control. #p < 0.05 vs. PH-Control (Ronco, et al. 2007).") Figure 4.
Figure 4.Immnublottintg analysis of VEGF protein expression in total liver lysates fraction at 72 h post-surgery. A. Line 1: Sh-Control, a midline laparotomy with liver manipulation was carried out on surgical sham control rats. Line 2: PHControl, two-thirds hepatectomy was performed to control rats. Line 3: PH-SNP1, twothirds hepatectomy was performed to rats that had received sodium nitropruside 0.25 mg/kg body weight, intravenously. B. Line 1: Sh-Control. Line 2: PHControl. Line 3: PH-AG, twothirds hepatectomy was performed to rats that had received aminoguanidine (AG) 100 mg/kg body weight, intraperitoneally. *p < 0.05 vs. Sh-Control. #p < 0.05 vs. PH-Control (Ronco, et al. 2007).
(0.07MB).What is the role of NO in the proliferative process? To answer this question, we analyzed how modification of NO levels modulate the proliferative process that takes place after PH. The results obtained by immunohistochemistry detection of proliferation cellular nuclear antigen (PCNA) indicate that no change occurs in the proliferation of the remaining liver under an increase of over 100% of NO. Nevertheless, an increase of NO lower than 35% enhances the proliferation index (Ronco, et al., unpublished data), suggesting that a moderate increase of NO improve the proliferative process at early stages of liver regeneration.
As we previously described, the specific role of NO in the balance of apoptosis/proliferation remains controversial considering the dual action of NO in either promoting or impairing programmed cell death. Analyzing our own results focusing on the action of NO in the regulation of apoptosis/survival proteins in liver regeneration, we observed an expression of survival factors when NO is in “small concentrations”, and an expression of pro-apoptotic proteins when NO is at “high concentrations”.
Figure 3 shows a schematic description of the regulation of apoptosis/proliferation balance after PH, mediated by the impact of increases in NO on anti-apoptotic proteins Bcl-xL and pro-apoptotic Bax.
- ø
NO and Vascular Endothelial Growth Factor (VEGF). One of the well-characterized functions of NO is as a mediator of vascular dilatation and permeability, as well as in vascular remodeling.2,8 Angiogenesis, i.e. the formation of new blood vessels, is a complex process that involves proliferation and migration of endothelial cells. This phenomenon is required for remodeling liver architecture following liver resection.67-69 The initial wave of hepatocyte proliferation is followed by endothelial cell proliferation and penetration of vascular hepatocellular islands leading to the formation of newly-formed sinusoids.70 While angiogenesis has been extensively studied in various diseases, particularly in carcinogenesis, its impact on physiological processes is less well known. In particular, its role in the physiological control of organ mass is still not completely understood. Information gathered from immunohistochemical studies points towards well-known angiogenic factors such as vascular endothelial growth factor (VEGF), one of the most potent angiogenic factors, as a main mediator of endothelial cell proliferation in the regenerating liver.68,71,72
After PH, both hepatocytes and nonparenchymal cells express VEGF mRNA, suggesting that VEGF plays a significant role in this process. Hepatocellular production of VEGF shows the maximal levels between 48 and 72 h after PH.71,73 An increase of VEGF production by hepatocytes correlates with an increase in VEGF receptor expression on endothelial cells after PH. Moreover, recent studies have shown that inhibition of angiogenesis with angiostatin impairs liver regeneration. Furthermore, an increased expression of VEGF and its receptors induce the proliferation of endothelial cells.71,74
It is known that NO plays an important role in the processes of vascularization, angiogenesis and permeabilization of tissue. Although there is a growing body of evidence that NO has angiogenic effects, partly mediated by VEGF, there is no unanimity of opinion on this regard.75,76 The effects of NO are greatly dependent upon cell type, cellular redox status, and the amount and chemical nature of NO donors.21 In previous studies, we showed that treatment of PH-animals with low doses of the direct NO donor SNP (0.25 mg/kg body wt) increases VEGF levels compared with PH-alone, and that the inhibition of NO synthesis (with the selective inhibitor of iNOS amino-guanidine) decreases VEGF protein levels, suggesting that NO is implicated in VEGF expression (Figure 4). Accordingly with ours results, other authors have reported that the exogenous addition of NO donors or increased levels of endogenous NO enhanced VEGF synthesis in rat vascular smooth muscle cells.58 Furthermore, in the rabbit cornea model of angiogenesis, VEGF-induced angiogenesis is blocked by L-NAME (non-selective NOS inhibitor), demonstrating that neovascularization is suppressed by the blockade of NO production.77 Besides, Taniguchi, et al. have shown that VEGF expression in regenerating rat liver occurs predominantly in periportal hepatocytes.73 They also demonstrated that VEGF is involved in proliferation of hepatocytes associated with proliferation of sinusoidal endothelial cells after PH in rats. Histological studies revealed that NO donor treatment increases the number of vascular structures in portal areas in PH animals and that aminoguanidine treatment reduces this rate,78 suggesting that the augmentation of NO levels increases periportal vascularization, probably via VEGF. In agreement with other authors, these studies provide further evidence that VEGF production is regulated by NO, and that this compound plays a central role in rat liver regeneration after PH.75,76 In our studies, we showed that the modification of NO levels at 5 h post-PH produces changes in VEGF protein levels at 72 h after PH.78 These results suggest that the NO increase during early steps of liver regeneration initiates an adaptive response leading to activation of transcriptional factors that act as signal transducers between cytoplasm and nucleus, which results in the regulation of VEGF expression.
- ø
 and the treatment with NO donor SNP2 (2.50 mg/mL) produce an over-increase of 100% of NO, leading to an increase of pro-apoptotic proteins Bax and p53, leading to apoptosis. Treatment with a NO donor, SNP1 (0.25 mg/mL), produces an increase lower than 35% of NO, thus leading to an increase of anti-apoptotic protein BclxL, which improves liver regeneration (⊥ not difference, ↑ increase, ↓ decrease).")
 100 mg/kg body weight, intraperitoneally. *p < 0.05 vs. Sh-Control. #p < 0.05 vs. PH-Control (Ronco, et al. 2007).")
Data revised and presented in this review lead to the assumption that NO activates, as a signaling molecule, different cellular mechanisms that can promote either cell growth or cell death. NO plays an important and diverse role during liver regeneration, with the potential to be either anti-or pro-apoptotic. Experimental evidence shows that the expression of iNOS is induced at early stages of liver regeneration after PH, provided a high amount of NO is present. When conditions are right for peroxinitrite generation, NO, via the formation of peroxynitrite, can damage cellular components such as mitochondria, leading to the opening of the membrane transition pores (MTP) with the consequent release of cytochrome c into the cytoplasm. In addition, NO can increase the expression of the pro-apoptotic proteins Bax and p53, resulting in cell death.
At early stages of liver regeneration, the small amounts of NO produced appear to be necessary and perhaps sufficient to produce an increase in anti-apoptotic protein Bcl-xL, which results in protection from apoptotic cell death. Also, this level of NO ensures a maximum increase of VEGF at 72 h post-hepatectomy, which is necessary to maintain sinusoidal perfusion and induce neovascularization.
This review highlights the regulating role of NO at early stages of liver regeneration, where NO is overproduced as a consequence of iNOS activation. Interestingly, in our studies, we observed that an increase of 35% of NO during the early stages of PH leads to an improvement of liver regeneration. Drugs that can modulate NO levels have been widely used as therapeutic agents in order to a favorable adjustment of the vascular environment. This offers the possibility of using these drugs, or even the designing new, hepatotropic drugs that regulate NO levels with the aim of improving the process of liver regeneration.
AcknowledgmentsThis work was supported by research grants from ANPCyT (PICT N° 32413, C.E. Carnovale PhD.) and from CONICET (PIP N° 5531, C.E. Carnovale PhD.).



