








Background: Gallbladder carcinoma (GBC) is a frequent neoplasm in Hispanic and native American populations. GBC is preceded by gallstones, chronic cholecystitis and dysplastic changes of the gallbladder epithelium. The knowledge of the molecular events involved in its pathogenesis is scarce. Aims: We investigated the role of TP53 inactivation in the sequential pathogenesis of GBC. Methods: Invasive tumor-, dysplastic-and histologically normal GB epithelial-cells were obtained from archival formalin-fixed tissues from GBC and GB from gallstone patients without GBC. Normal GB epithelia from 5 non-gallstone specimens were also studied. DNA extracted was examined for loss of heterozygosity (LOH) using 2 microsatellite markers and for TP53 mutations at exons 5 to 8. Results: GBCs demonstrated a high frequency of LOH (81%) and mutation (67%), and both abnormalities indicating gene inactivation were detected in 52%. Similar frequency of TP53 abnormalities and gene inactivation (38%) were detected in their accompanying normal and dysplastic epithelia. Noteworthy, one third of normal and dysplastic epithelia obtained from GBs of gallstone patients without GBC demonstrated either TP53 allele loss or mutation, but gene inactivation was less frequent (11%). Most mutations affected exons 5 and 7, and they were more frequently missense point mutations. The same TP53 mutation was detected in only a subset (27%) of comparisons between non-malignant epithelia adjacent to GBCs, indicating that TP53 mutation occurs independently at several epithelial foci. Conclusions: These findings indicate that TP53 abnormalities are early and frequent events in the pathogenesis of GBC, starting from chronic cholecystitis.
§ Supported by Grants FONDECYT (Fondo Nacional de Desarrollo Científico y Tecnológico) #1040820, #1971100, and # 1990489.
Abbreviations: GBC, gallbladder carcinoma; LOH, loss of heterozygosity; TSG, tumor suppressor gene.
IntroductionGallbladder carcinoma (GBC) is a relatively uncommon neoplasm in the world, but there is considerable geographic variation in its incidence.1 In Hispanic populations with native American background, GBC is one of the most frequent neoplasms, where it is the leading cause of cancer deaths in females.1 Although GBC has been associated with genetic and environmental risk factors, there is limited information about the molecular changes involved in its pathogenesis.2 It has been claimed that cholesterol gallstone disease and subsequent chronic aseptic cholecystitis is the most relevant risk factor for GBC.13 In fact, prevalence of GBC correlates with the prevalence of gallstone disease in different populations.1 As with other epithelial neoplasms, it has been well established that invasive GBC is preceded by preneoplastic lesions, including dysplastic changes of the gallbladder epithelium.4
Multiple genetic changes are associated with the development of many human cancers, and molecular studies in several neoplasms have demonstrated their association with the activation of dominant oncogenes and inactivation of recessive tumor suppressor genes. The TP53 tumor suppressor gene, located on the short arm of the chromosome 17 (17p13.1), encodes a 53-kD nuclear phosphoprotein that acts as a “master” transcriptor factor.5 p53 is believed to play a major role in maintaining the integrity of the genome since loss of p53 function allows inappropriate survival of genetically damaged cells, leading to the evolution of a cancer cell.5 Mutations of the TP53 gene are among the most frequent genetic alterations in human cancers and are thought to play a role in the pathogenesis of many malignancies.6,7 One copy of the chromosomal region 17p13.1 which houses TP53 gene is frequently deleted, and subsequent mutational in-activation of the remaining allele occurs.7
As with several other human neoplasms, TP53 mutations,8-14 allele loss1,15 and immunohistochemical protein over-expression16-22 have been reported as frequent events in advance GBC. Analyses of exon 5 to 8 of the TP53 gene have demonstrated point mutations in 31 to 70% of gallbladder carcinomas, and no particular hot-spot has been identified so far.8-13 Our allelotyping studies on GBC have indicated that allelic loss or loss of heterozygosity (LOH) at the TP53 locus is a frequent (91%) molecular change detected in this neoplasm.1,15 Although several studies have demonstrated that TP53 mutation or allele loss are common events in advanced GBC, there is no study addressing both abnormalities responsible for the gene inactivation in GBC and in clinical steps preceding GBC, such as gallbladder epithelium from gallstone patients with chronic cholecystitis. Thus, the present study was undertaken to study the role of TP53 gene inactivation abnormalities in the sequential pathogenesis of GBC by examining histologically normal gallbladder epithelia and preneoplastic lesions from gallstone patients without GBC, and in patients with invasive GBC.
Materials and methodsArchival Tumor Specimens. Formalin-fixed paraffin-embedded material from 30 surgically resected primary invasive gallbladder carcinomas was obtained from cholecystectomy specimens resected between 1996 and 2000 at the Catholic University Medical School Hospital, Santiago, Chile, as part of an Institutional Review Board approved study. The patients consisted of 20 women and 10 men ranging in age from 48-79 years (mean age, 57 years). Seven (23%) were well differentiated, ten (33%) were moderately differentiated, and 13 (43%) were poorly differentiated tubulo-papillary adenocarcinomas. The majority of the tumors were advanced GBCs (25 cases, 83%) with invasion of the gallbladder serosa; the remaining were early GBCs, with invasion of the submucosa (3 cases, 10%) or muscularis propia (2 cases, 7%) of the gallbladder.
Normal Epithelium and Preneoplastic Lesions Accompanying Gallbladder Cancer. Twenty-three histologically discrete foci of non-invasive gallbladder epithelia were identified adjacent to 16 GBCs, each consisting of at least 1,000 cells. These included 7 histologically normal epithelia and 16 dysplasias. The dysplastic lesions were scored using published criteria for their histopathological identification in the gallbladder epithelium.4
Normal Epithelium and Preneoplastic Lesions from Patients without Gallbladder Cancer. Histologically normal and dysplastic epithelia from 61 gallbladder specimens obtained from gallstone patients with chronic cholecystitis in the absence of GBC were also selected. These included 39 gallbladder specimens with discrete epithelial dysplastic foci, and 22 with histologically normal epithelia. In the dysplastic and non-dysplastic cases, the whole gallbladder specimen was histologically examined to rule out the presence of invasive carcinoma and dysplasia, respectively. In addition, five normal gallbladder specimens were obtained from non-gallstone patients and their histologically normal epithelium was examined.
Microdissection and DNA Extraction. Serial 5 μm-sections were cut from archival, formalin-fixed, paraffin embedded tissue. All slides were stained with hematoxylineosin, and one of the slides was coverslipped. The coverslipped slide was used as a guide to localize regions of interest for microdissection for the other slide. Laser capture microdissection and DNA extraction were performed from precisely identified areas of stromal lymphocytes, invasive carcinoma and dysplastic or normal epithelia from gallbladder specimens as previously described from non-coverslipped hematoxylin-eosin stained slides.23
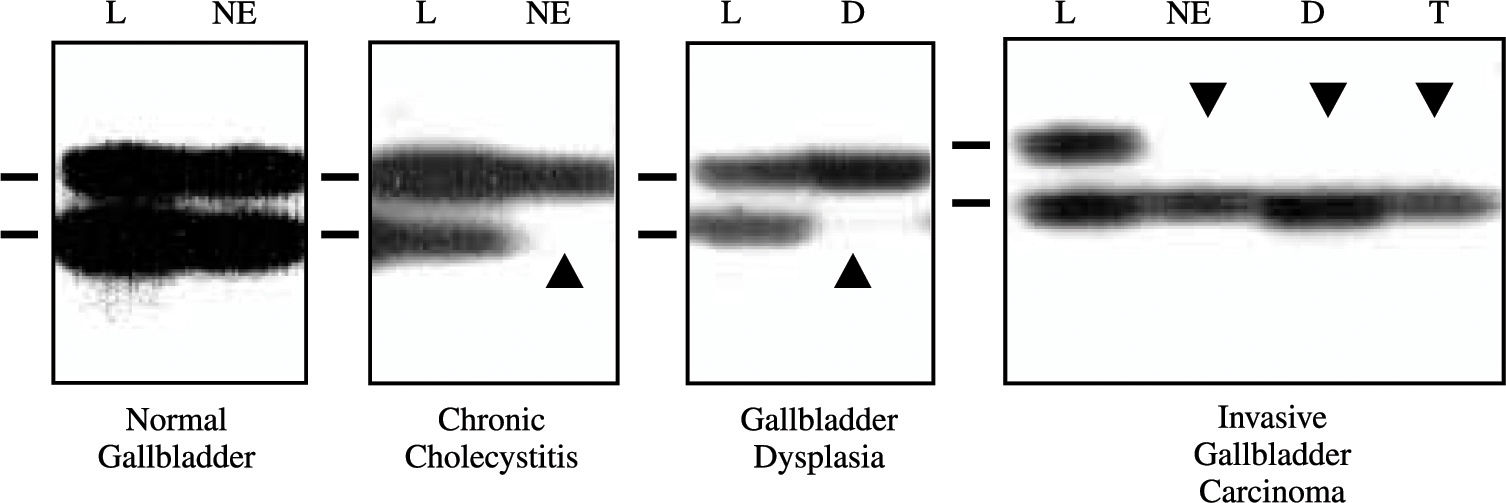
Loss of Heterozygosity Analysis of TP53 Locus. Extracted DNAs were utilized for loss of heterozygosity (LOH) analysis at TP53 locus (17p13.1) using two polymorphic microsatellite markers, a dinucleotide CA repeat flanking the gene24 and a pentanucleotide AAAAT repeat located in intron 1 of the gene.25 A two-round PCR strategy (multiplex PCR followed by uniplex PCR) was utilized to amplify each marker, as described previously.23 The final product was separated on a 6% denaturing poly-acrylamide gel and subjected to autoradiography. LOH was scored by visual detection of complete absence of one allele of informative cases (Figure 1).

Representative autora-diographs of microsatellite analysis for allele loss using the pen-tanucleotide repeat on the TP53 locus in gallbladder specimens. L, lymphocytes; NE, normal epithelium; D, dysplasia; and, T, tumor. Horizontal bars on the left of each autoradiograph indicate the main allelic band. Closed arrowheads indicate allele losses.
TP53 Mutation Analysis. Mutations were examined in exons 5 to 8 which harbor 87% of gene mutations reported for TP53 in human neoplasms.6 These exons encode for the critical region which determined p53 conformation, specific binding to DNA consensus sequences, and sequence-specific transcriptional activity which may be essential for growth suppression.6 We screened for mutations using nested PCR methodology and non-isotopic RNase cleavage assay (NIRCATM) Mismatch Detect II™ kit (Ambion, Austin, TX, US), followed by direct sequencing of both strands of abnormal bands at the NIR-CA assay (Figures 2A, B). Specificity of indetified point mutations were always confirmed by a second sequencing analysis starting from the original DNA samples of tumoror epithelial-cells and corresponding stromal cells. The NIRCATM technique has been adapted from a screening method based on the observation that RNase A is frequently able to cleave a single unpaired base (mismatch) in an RNA probe hybridized to an experimental target containing a point mutation.26,27 Briefly, in the NIRCA™ procedure the target substrates for RNase digestions are RNA/RNA duplexes made by hybridizing complementary wild and experimental transcripts. The outline of the NIRCA™ procedure can be broken down in the five following steps: 1) PCR amplification of experimental and wild type control DNA sequences. 2) Transcription of both strands of experimental and wild type PCR product. 3) Hybridization of complementary experimental and wild type transcripts. 4) Digestion of RNA duplexes with RNases (RNase 1, RNase T1 and RNase A). 5) Separations of digestion products by gel electrophoresis in agarose. Primers and PCR conditions to amplify exons 5 to 8 (step 1) from formalin fixed and paraffin embedded tissues have been previously published by some of us.28 Step 2 and 5 were performed according to manufacturer’s conditions (Mismatch Detect IITM kit, Ambion, Austin, TX, US). The corresponding positive (lung cancer cell lines H2106, H23, H460) and negative (normal lymphocytes) controls were included in the analysis for each exon. NIRCATM assay was able to identify point mutations in all cell lines in different experiments and were consistently negative for mutations in negative control samples (stromal lymphocytes), showing a high sensitivity and specificity.
, using PCR amplification, non-isotopic RNase cleavage assay and agarose gel electrophoresis (four gels, Panel A), followed by sequencing (Panel B). Panel A, Open arrowheads indicate cases with abnormal bands selected for sequencing analysis. M, size marker; C, positive controls (lung cancer cell lines having TP53 mutations); S, negative controls, stromal cells; and, T, tumor cases. Panel B, sequencing of exon 5 showing a point mutation in codon 164 in invasive carcinoma sample.")
Representative example of the PCR-based TP53 mutations screening method (exons 5 to 8), using PCR amplification, non-isotopic RNase cleavage assay and agarose gel electrophoresis (four gels, Panel A), followed by sequencing (Panel B). Panel A, Open arrowheads indicate cases with abnormal bands selected for sequencing analysis. M, size marker; C, positive controls (lung cancer cell lines having TP53 mutations); S, negative controls, stromal cells; and, T, tumor cases. Panel B, sequencing of exon 5 showing a point mutation in codon 164 in invasive carcinoma sample.
Data Analysis. Statistical analysis was performed using chi-square and Fisher Exact tests. The cumulative binomial test was used to examine the likelihood that the occurrence of a particular event (loss of the same allele in the invasive carcinoma and an associated epithelial sample) occurs at a particular probability when observed in repeated trials. When the results are compared with a chance occurrence or nonoccurrence, the particular probability of comparison is 0.5. Probability values of P < 0.05 were regarded as statistically significant.
ResultsTP53 Abnormalities in Invasive GBC. LOH at the TP53 locus using two microsatellite markers (81%), and gene mutation at exons 5 to 8 (67%) were frequently detected in invasive GBC (Table I). Any TP53 abnormality was found in most (95%) tumors. GBC demonstrated a high frequency of multiple TP53 mutations in exons 5 to 8, with 15 out of 20 (75%) tumors having more than one mutation and the mean number of mutations detected by case was 3 (range 1 to 5) (data not shown). As TP53 allele loss and mutation analyses were performed in the same tumor samples, we examined in GBCs the frequency of gene inactivation defined by the presence of both abnormalities in the same tumor specimen (Table I). Twenty-one GBCs examined for mutation were informative for LOH at any microsatellite marker tested, and of those 11 (52%) tumors demonstrated both TP53 changes. For TP53 inactivation analysis, only mutations producing amino acid change (nonsense, frame-shift and missense) were considered. No correlation between TP53 changes and clinico-pathological features of GBC was detected (data not shown).
Frequency of TP53 abnormalities (LOH, mutation and any change) and gene inactivation in the sequential pathogenesis of GBC.
| Histology N (%)1 | Loss of Heterozygosity N (%)2 | Mutation N (%)3 | Any Change N (%)4 | Gene Inactivation |
|---|---|---|---|---|
| Normal Gallbladder | 0/4 | 0/5 | 0/4 | 0/4 |
| Chronic Cholecystitis Specimens5 | 16/46 (35%) | 15/61 (25%) | 26/46 (57%) | 5/46 (11%) |
| Normal Epithelium | 6/17 (35%) | 6/22 (27%) | 11/17 (65%) | 1/17 (6%) |
| Dysplasia | 10/29 (35%) | 9/39 (23%) | 15/29 (52%) | 4/29 (14%) |
| Cancer Specimens6 | 28/34 (82%) | 35/53 (66%) | 32/34 (96%) | 16/34 (47%) |
| Normal Epithelium | 2/3 (67%) | 4/7 (57%) | 3/3 (100%) | 1/3 (33%) |
| Dysplasia | 9/10 (90%) | 11/16 (69%) | 9/10 (90%) | 4/10 (40%) |
| Invasive Carcinoma | 17/21 (81%) | 20/30 (67%) | 20/21 (95%) | 11/21 (52%) |
| Total | 44/84 (52%) | 50/119 (42%) | 58/84 (69%) | 21/84 (25%) |
TP53 Abnormalities in Preneoplastic and Histologically Normal Gallbladder Epithelia. To investigate the role of TP53 abnormalities in the sequential pathogenesis of GBC, histologically normal and dysplastic epithelia accompanying GBCs and in chronic cholecystitis from gallstone patients without GBC were examined. Although a limited number of histologically normal epithelia and dysplasias accompanying invasive GBC were studied, the majority of those specimens demonstrated a high incidence of both allele loss (normal epithelium 67%, and dysplasia 90%) and mutation (normal epithelium 57%, and dysplasia 69%) (Table I). However, all the epithelial specimens studied for allele loss and mutation were obtained exclusively from invasive GBC having the corresponding TP53 changes. Somewhat striking, approximately one third of the histologically normal and dysplastic epithelia samples obtained from gallstone patients with chronic cholecystitis and without GBC demonstrated either TP53 allele loss (normal epithelium and dysplasia 35%) or mutation (normal epithelium 27%, and dysplasia 23%) (Table I). While any TP53 change was found in almost all normal and dysplastic samples associated to GBCs, they were detected in nearly half of the epithelia from chronic cholecystitis in the absence of GBC. In contrast, normal epithelium obtained from five histologically normal gallbladders from patients without gallstones did not show any TP53 change (Table I).
A progression in the number of TP53 mutations by sample according with increasing histopathological grade was observed (data not shown). As their corresponding tumors, histologically normal and dysplastic epithelia associated to GBC demonstrated frequently multiple TP53 mutations. While normal epithelium from chronic cholecystitis presented a single mutation for case, dysplasias obtained from the same type of non-malignant gallbladder specimens demonstrated intermediate number (mean 1.69; range 1 to 2) of mutations.
Gene inactivation phenomenon by examination of both TP53 abnormalities was more frequently detected in histologically normal and dysplastic epithelia from gallbladder specimens with invasive GBC than those obtained from chronic cholecystitis (Table I). While 1 out of 3 (33%) normal and 4 out of 10 (40%) dysplastic epithelia accompanying GBCs demonstrated TP53 inactivation, this phenomenon was present in 1 out of 17 (6%) normal epithelium and 4 out of 29 (14%) dysplasias obtained from chronic cholecystitis without GBC.
TP53 Mutation Pattern in the Pathogenesis of GBC. The mutation distribution by exons and codons detected in invasive GBC, preneoplastic lesions and histologically normal epithelium is shown in figure 3. Overall, ninety-five TP53 mutations were detected in GBC and epithelial samples, and tumor and dysplastic samples demonstrated at very high frequencies more than one mutation. Mutations on tumors (17 out of 46 mutations; 37%) and their accompanying dysplastic and normal epithelia (18 out of 30; 60%) affected more frequently to exon 5, and those detected in epithelial samples obtained from chronic cholecystitis specimens (10 out of 19; 53%) were more frequently found in exon 7. The distribution of mutations by codon showed no particular hot-spot in gallbladder samples; however, the codons showing more frequently mutations were 144 (N = 11) and 248 (N = 6) figure 3.
 distribution (N = 94) by codons in GBC pathogenesis. Boxes indicate exons. The distribution of mutations by codon showed no particular hot-spot in gallbladder samples; however, the codons showing more frequently mutations were 144 (N = 11) and 248 (N = 6).")
TP53 point mutation (vertical lines) distribution (N = 94) by codons in GBC pathogenesis. Boxes indicate exons. The distribution of mutations by codon showed no particular hot-spot in gallbladder samples; however, the codons showing more frequently mutations were 144 (N = 11) and 248 (N = 6).
Twenty-five percent (24 out of 95) of the TP53 mutations detected were silent mutations (Figure 4, A), and they were not detected in the corresponding stromal tissue obtained from the same patient confirming that they were not polymorphism. Nearly half of the mutations detected in normal (3 out of 6; 50%) and dysplastic (6 out of 13; 46%) epithelia from chronic cholecystitis specimens were silent. These frequencies were significantly higher (P < 0.015) than the incidences of silent mutations in tumors (12 out of 46; 26%) and their accompanying normal (1 out of 7; 14%) and dysplastic (2 out of 23; 9%) epithelia. Among the mutations producing amino acid change (N = 71 mutations), missense point mutations (N = 57; 80%) were the commonest observed (Figure 4, A). Nonsense point mutations were less frequent (N = 13; 18%), deletion producing frame-shift mutation were extremely rare (N = 1; 2%), and no insertion was detected.
 type frequencies in GBC pathogenesis.")
By far, transition type of TP53 point mutations (77 out of 94; 82%) showed the greatest incidence in GBCs, dysplastic lesions and normal epithelia. It was the only type detected in normal and dysplastic samples obtained from chronic cholecystitis. Of great interest, transitions G:C>A:T were the commonest mutation detected (68 out of 94; 72%) in our study, followed by A:T>G:C change (9 out of 94; 10%; (Figure 4, B).
TP53 Allele Specific Loss and Mutation. Previous studies in GBC and other neoplasms demonstrated that at any one locus, loss of parental alleles was not random, and that there was a strong tendency for the identical allele to be lost in all non-neoplastic and neoplastic foci examined.15,29 We refer to this phenomenon as allele specific loss. We determined the frequency of allele specific loss in the 16 epithelial and tumor foci demonstrating one or more sites of allelic loss in 8 GBC cases. The same parental allele was lost in all 13 comparisons involving both microsatellite markers examined at the TP53 locus. The possibility that this occurred by chance alone is remote as tested by the cumulative binomial test (P = 0.000012). Then, we examined the TP53 mutation pattern to mark the different foci of cancer and accompanying gallbladder epithelia in individual cases. Of interest, the same TP53 mutation compared to the corresponding tumor was detected in only 4 out 11 (27%) normal and dysplastic epithelia. Thus, while the allele specific loss phenomenon was detected comparing our invasive tumors and their corresponding normal and dysplastic epithelia, they did not demonstrate TP53 specific mutations.
TP53 Abnormality Patterns. To determine the sequence of changes involved in the inactivation of TP53 in the pathogenesis of GBC, we analyzed the pattern of allele loss and mutation detected in tumors and normal and dysplastic epithelia obtained from malignant and non-malignant gallbladder specimens. Seventy-two tumor and epithelial samples informative for any microsatellite marker used to test LOH at TP53 locus were examined for gene mutations, and the 4 possible patterns of TP53 abnormalities (I, no change; II, only LOH; III, only mutation; and, IV, LOH and mutation) were detected. The relationship between histopathological diagnoses and patterns of TP53 inactivation is displayed in figure 5. Of interest, pattern II (only LOH) was more frequently detected than pattern III (only mutation) in overall samples (22/52, 42%, vs 11/52, 11%), tumors (7/20, 35%, vs 4/20, 20%) and epithelia obtained from malignant and non-malignant specimens (15/32, 47%, vs 7/32, 22%). Statistically significant differences were detected only when patterns of overall and epithelial samples were compared (P = 0.017, and P = 0.39, respectively). For this type of analysis, only mutations producing amino acid (non-silent mutations) changes were considered.
 observed in 52 non-malignant and cancer gallbladder specimens. Relationship between histological categories and patterns of TP53 abnormalities. Normal and dysplastic epithelia from non-malignant gallbladder specimens were obtained from chronic cholecystitis. Gene inactivation (pattern IV) is seen mostly in cancer specimens. Note that pattern II (only LOH) was more frequently detected than pattern III (only mutation) in overall samples, tumors and epithelia obtained from chronic cholecystitis.")
Patterns of TP53 abnormalities (LOH and non-silent mutation) observed in 52 non-malignant and cancer gallbladder specimens. Relationship between histological categories and patterns of TP53 abnormalities. Normal and dysplastic epithelia from non-malignant gallbladder specimens were obtained from chronic cholecystitis. Gene inactivation (pattern IV) is seen mostly in cancer specimens. Note that pattern II (only LOH) was more frequently detected than pattern III (only mutation) in overall samples, tumors and epithelia obtained from chronic cholecystitis.
The presence of a single TP53 abnormality (LOH or point mutation) was detected in the majority of GBCs and in nearly half of them demonstrated both changes in the same sample consistent with inactivation of this tumor suppressor gene. Our data are in agreement with the high frequency of TP53 abnormalities previously reported by us and others in GBC obtained from patients from Chile13-5 and elsewhere.8-13 However, there is no study addressing both abnormalities responsible for the TP53 inactivation in invasive tumors and epithelia involved in the sequential pathogenesis of this highly malignant tumor.
In several neoplasms it has been established that multiple sequential genetic changes are associated with the development of invasive tumors. To investigate the role of TP53 abnormalities in the sequential pathogenesis of GBC, histologically normal and dysplastic epithelia obtained from GBC and also from chronic cholecystitis gallbladder specimens obtained from gallstone patients without GBC were examined. Although a limited number of epithelial samples accompanying invasive tumors were studied, our finding of high frequencies of TP53 abnormalities and gene inactivation in normal and dysplastic gallbladder epithelia associated to GBC confirmed that this gene plays an important and early role in the sequential pathogenesis of this tumor. Although a high incidence of allelic loss at TP53 locus occurring early during the sequential pathogenesis of GBC have been previously reported by us,15 this is the first report on TP53 mutation and gene inactivation phenomenon in normal and dysplastic gallbladder epithelia obtained from GBC and chronic cholecystitis without GBC.
One of the most striking findings of our study is related to the analysis of TP53 abnormalities in chronic cholecystitis epithelia obtained from gallstone patients without GBC. We demonstrate for the first time that approximately one third of the histologically normal and dysplastic epithelia obtained from those samples have either TP53 allele loss or mutation, and any change was detected in nearly half of the samples. These findings lend stronger support to the view of TP53 as an important and early event in gallbladder tumorigenesis. Moreover, normal epithelium obtained from 5 normal gallbladder specimens available to us without gallstones and inflammatory changes did not demonstrate any TP53 abnormality. The development of epithelial cancers requires multiple mutations, and the stepwise accumulation of these mutations may represent an inherent mutator phenotype.30 Thus, we speculate that is likely that those normal and dysplastic epithelia from chronic cholecystitis that have accumulated multiple mutations, including TP53 abnormalities, are at higher risk for progression to GBC. The morphologically defined stages in which TP53 mutation appears in the sequential development of other epithelial tumors have been established,6 and there are few examples of human tissues with frequent TP53 mutation occurring in normal epithelium from patients without neoplasm.31-33
Tumor suppressor genes are believed to be inactivated via a two-step process involving both alleles, including among other mechanism mutation, deletion and aberrant methylation. For TP53, one copy of the chromosomal region 17p13.1 is frequently deleted and mutational inactivation of the remaining allele occurs.7 By examining TP53 locus allele loss and synchronic gene mutation in the same tumor and epithelium samples from malignant and non-malignant gallbladder specimens, we have provided for the first time data on the role of TP53 inactivation phenomenon in the sequential pathogenesis of GBC. Interestingly, TP53 inactivation was detected in nearly half of invasive tumors, and less frequently in dysplastic and normal epithelia accompanying GBCs. But such phenomenon was also detected in few normal (6%) and dysplastic (14%) epithelia from chronic cholecystitis specimens in the absence of tumor. We hypothesize that those normal and dysplastic lesions from gallstone patients having TP53 inactivation are at higher risk for malignant transformation, and additional genetic change are required to progress to atypical lesion.
The sequence of molecular events involved in TP53 inactivation has not been demonstrated for sporadic cancers. To determine the sequence of changes involved in the inactivation of TP53 in the pathogenesis of GBC, we analyzed the pattern of allele loss and mutation detected in tumors and in normal and dysplastic epithelia obtained from malignant and non-malignant gallbladder specimens. Our finding of allele loss as a single TP53 abnormality significantly more frequent compared to mutations in gallbladder tumor and epithelium samples suggest that allele loss at TP53 locus may usually precede gene mutation. It may be possible that loss of one allele destabilizes and predisposes to mutation of the remaining allele.
The same parental TP53 allele was lost in 100% of comparisons in non-neoplastic lesions as in the corresponding invasive carcinomas. We refer to this phenomenon as allele specific loss, and we have previously documented this phenomenon in GBC.15,29 Although the mechanism underlying allele specific loss remains unknown, recently we have demonstrated in GBC that this phenomenon is not related to a clonal relationship between the epithelial foci and their corresponding tumors using allele loss and microsatellite instability pattern analyses.29 Of interest, the same TP53 mutation was detected in only a subset (27%) of comparisons between non-malignant epithelia and corresponding GBCs, indicating that in gallbladder malignant transformation TP53 mutations occur independently at several gallbladder epithelial foci, which are probably not clonally related which is also consistent with the LOH pattern described for GBC. These findings suggest that may be a strong field effect in gallbladder epithelium with chronic cholecystitis strongly predisposes to cancer.
The distribution of TP53 mutations in our GBCs was similar to the one previously reported in this tumor type, being more frequent at exon 5.8-13 In our cases, the distribution of mutations by codon showed no particular hotspot; however, codons 144 and 248 demonstrated more frequent mutations. Codon 248 is a hot-spot for mutations in human neoplasms (e.g., colon, skin, breast and lung), and encodes for an amino acid (Arg 248) which directly contact DNA and appears to be critical in stabilizing the protein-DNA interface.6 Most TP53 mutations detected in GBC and gallbladder epithelia were single base substitutions or point mutations followed by missense mutations. This pattern of mutations is consistent with previous reported data on GBC.8-14 Of interest, most of our point mutations were transitions (82%), especially G:C>A:T (72%) changes. Similar mutation pattern can be found in 74 GBCs registered in the TP53 mutation database maintained by the International Agency for Research on Cancer (IARC; version R9, July 2004; www.iarc.fr/p53).34
Transitions have been reported as the most frequent base substitutions on TP53 in human cancers35. Frequent transitions, especially G to A and C to T at CpG sites, are features of mutational TP53 spectra found in cancers not strongly linked to specific exogenous carcinogens6 and they are thought to be endogenous mutations caused by spontaneous deamination of 5-methylcytosine.36 Among 39 CpG dinucleotides in the human TP53 coding region, codons 175, 213, 245, 248, 273 and 282 are known as hot spots resulting from endogenous mutational processes, and mutations at 4 of those codons were detected in our cases.7 Thus, we speculate that endogenous carcinogens associated to the chronic inflammation present in cholesterol gallstone patients might be one of the most relevant initiating steps in triggering gallbladder carcinogenesis. It has been suggested that long-standing chronic inflammatory process and their oxidative stress products may be associated to malignant transformation by generating frequent and early TP53 changes in other gastrointestinal neoplasms, such as esophageal adenocarcinoma in Barrett’s esophagus37 and colon carcinoma in ulcerative colitis.38 Of great interest, like our gallbladder specimens, in both ulcerative colitis (> 50%) and Barrett’s esophagus (81%) their corresponding epithelia usually demonstrate frequent G:C>A:T transitions type of TP53 mutations.38
It has been established that aseptic or chemical chronic inflammation is involved in gallstone pathogenesis,39 and that the presence of a lithogenic bile (cholesterol supersaturated bile) is able to generate a permanent aseptic inflammatory response in the gallbladder wall.39,40 This inflammatory phenomenon, probably associated to other carcinogenic stimulus, might contribute to the development of the sequential molecular and phenotypic abnormalities that ends in GBC. Our finding of TP53 mutations early in the pathogenesis of GBC, including chronic cholecystitis, and the detection of a mutational pattern associated to endogenous mutational phenomenon suggests that chronic inflammatory process related to cholesterol gallstone may play an important role in TP53 abnormalities and GBC development.



