



Neuroendocrine tumors comprise approximately 1-2% of all gastrointestinal tumors, and while the liver is the most common site for metastasis of these tumors, primary hepatic neuroendocrine tumors are very rare entities. Since first being reported in 1958, there have been less than 150 cases reported in the literature. Because of the infrequent occurrence of these tumors, the pool of data available for analysis regarding these tumors is small. As such, the medical community must rely on the publication of case report data to further enlarge this data pool, with the hopes of eventually having enough data to draw meaningful, statistically significant conclusions with regard to diagnosis and management of these rare tumors. We have encountered two patients at our institution within the last year with primary hepatic neuroendocrine tumors. We present their cases in this manuscript in an effort to contribute to the available data on the disease. We also provide a concise review of the literature available to date regarding primary hepatic neuroendocrine tumors.
Neuroendocrine tumors (NETs) comprise approximately 1-2% of all gastrointestinal tumors, and while the liver is the most common site for metastasis of these tu-mors,1 primary hepatic neuroendocrine tumors (PHNETs) are very rare entities. The first case of such a tumor was reported was by Edmonson in 1958.2 Since then, there have been less than 150 cases reported in the literature, and they comprise approximately 0.3% of all neuroendocrine tumors.3 Because these tumors represent such a small minority of NETs as a whole, established algorithms for both diagnosis and management of these lesions do not exist. Survival rates for these tumors is very good, however, with 10 year survival reported as high as 73%.3 It is therefore important to accurately report cases of these tumors when they occur, so as to increase the total body of data available for subsequent analyses, allowing for better characterization of these tumors, and development of formalized diagnostic and treatment pathways. In this report, we present 2 PHNET cases and discuss diagnostic tools, surgical approach, and outcomes.
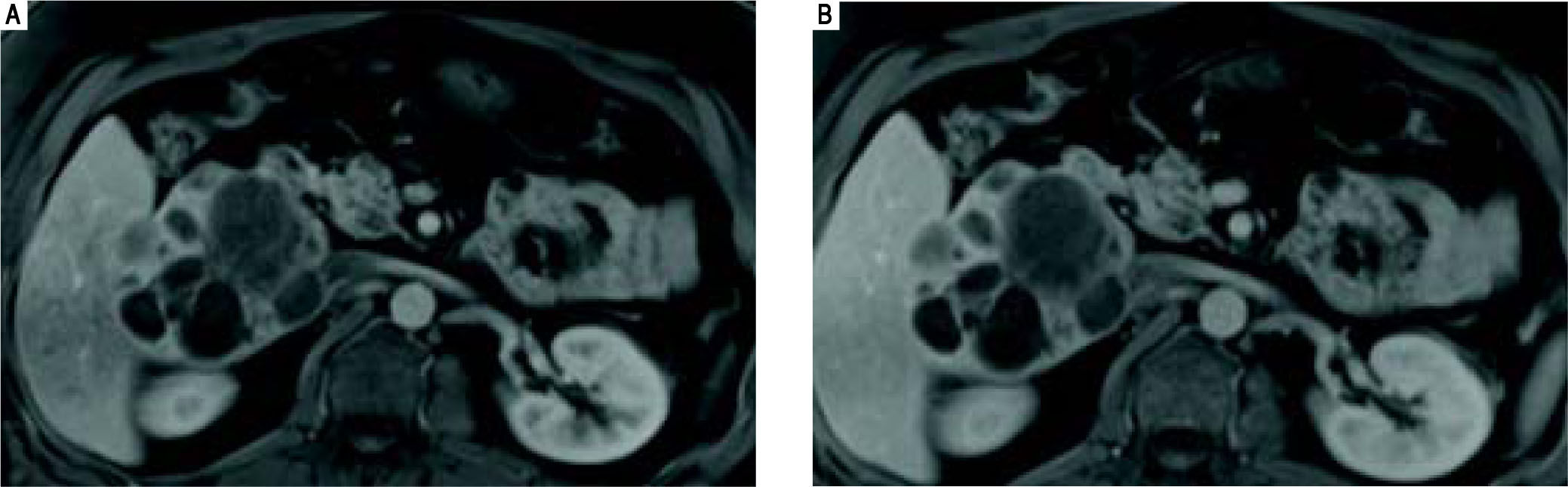
CASE REPORTCase 1Case 1 is a 41-year old male with a past medical history significant for severe aortic stenosis secondary to a congenital bicuspid aortic valve, non-ischemic cardiomyopathy and CHF (EF 25%), pulmonary hypertension, and laparoscopic cholecystectomy. As part of a preoperative work up for aortic valve replacement, he underwent a CT chest, which incidentally noted an 8.8 x 8.4 cm multiloculated lesion located within the caudate lobe of the liver (imaging not available). This was followed up with an MRI abdomen which redemonstrated a large 10.2 x 8.2 cm exophytic multiloculated complex cystic mass centered within the caudate lobe of the liver, with mild associated mass effect on adjacent portal vessels, common bile duct, IVC, and duodenum (Figure 1). The cystic locules of the mass appeared clustered together, with liver parenchyma insinuating itself between the adjacent cysts. Several of the cysts demonstrated fluid levels.
 and portal (B) venous phases demonstrates that the multiloculated mass does not arterial phase enhance, and its soft tissue components are isointense to the liver during the portal venous phase.")
Case 1. MRI abdomen. Axial fat-suppressed gadolinium-enhanced T1-weighted MR images obtained in the arterial (A) and portal (B) venous phases demonstrates that the multiloculated mass does not arterial phase enhance, and its soft tissue components are isointense to the liver during the portal venous phase.
He subsequently underwent uncomplicated aortic valve replacement with a St. Jude mechanical valve. His postoperative course was uncomplicated and he was discharged to home on Coumadin. Postoperatively, the patient was discussed multiple times by the institutional GI tumor board. Pelvic MRI was performed to rule out any primary lesion within the pelvis, and he was referred to surgery. His laboratory profile at that time showed very mild hyperbilirubinema (Tbil 0.92, Dbil 0.20) and an otherwise unremarkable liver profile, and the decision was made that the patient required resection of this mass, pending negative ecchinococcal serologies, which were subsequently negative. The patient's Coumadin was held and he was then taken to the operating room for an uncomplicated segment 1 liver resection. During the surgery, thorough exploration of the abdominal cavity was negative for any additional masses. His postoperative course was unremarkable except for development of a small 2 x 6 cm hematoma adjacent to the porta hepatis. Because of this, resumption of his anticoagulation was delayed. The patient was discharged to home on postoperative day 6 with instructions to resume his Coumadin as an outpatient.
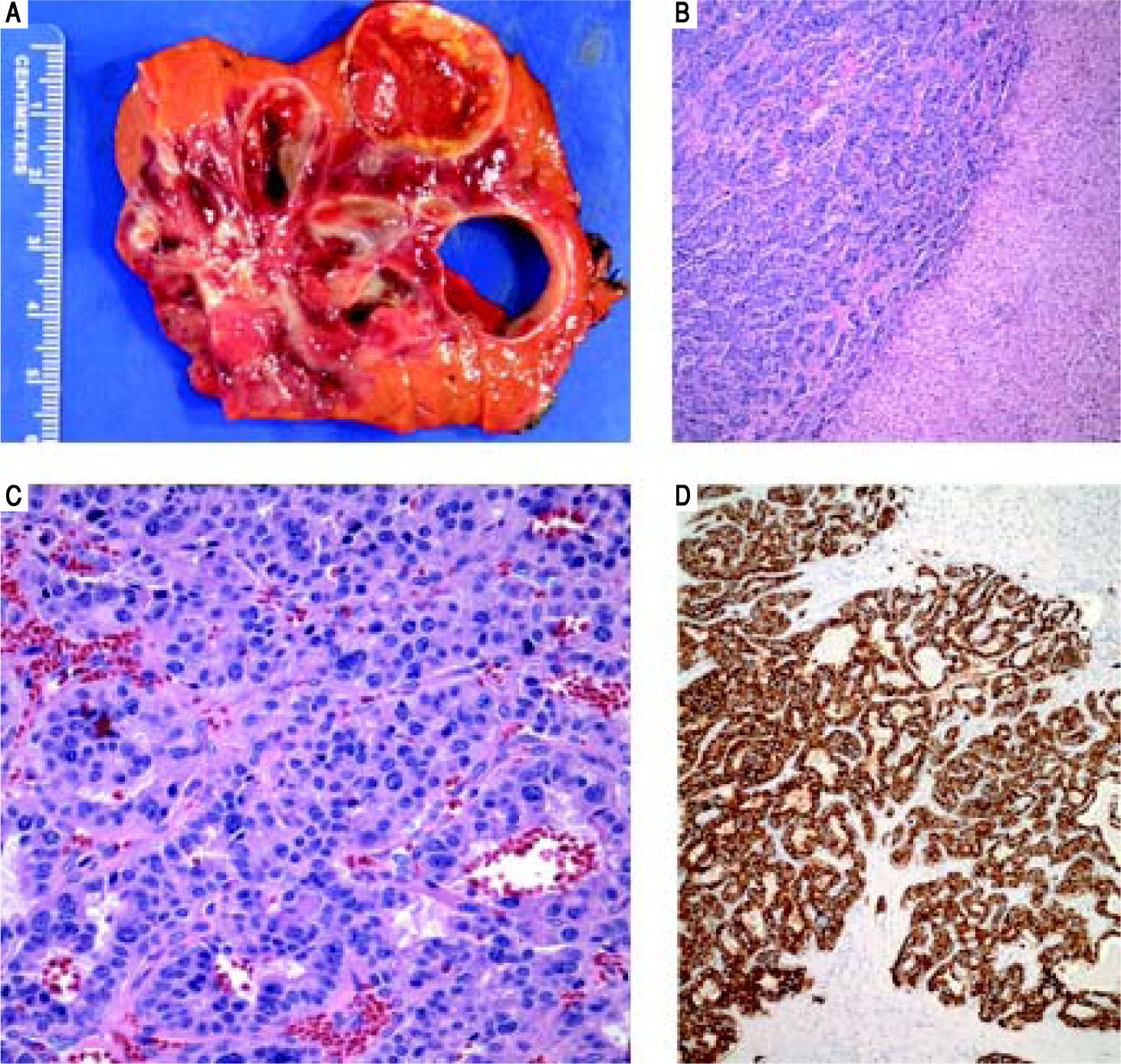
Final surgical pathology (Figure 2) showed a well differentiated neuroendocrine tumor, 11 cm in greatest dimension, with areas of cystic degeneration. There were up to 4 mitotic figures per 10 high power fields and the Ki-67 index was 6%, further classifying this as an intermediate grade lesion (WHO grade 2). Focal lymphovascular invasion was present. Immunohistochemically, the tumor cells were positive for neuroendocrine markers (CD56, chromogranin, and synaptophysin), MOC-31 and focally positive for CK7; whereas negative for hep par-1, glypican 3, polyclonal CEA, arginase-1, CK20, OCT4, pax 8 and CDX-2. The uninvolved liver was unremarkable and surgical margins were negative. An Octreotide Scan was performed 3 months postoperatively and was negative for any evidence of recurrent or persistent disease. Postoperative chromogranin A level was 27 ng/mL (ref.: 0-95 ng/mL) and his urine 5-HIAA was 2.1 mg/24 h (ref: < 6 mg/24 h), further suggesting that there is no persistent disease.
. C. On higher power, the tumor cells display round nuclei and moderate amounts of eosinophilic cytoplasm. Nuclei show a characteristic stippled (“salt and pepper”) chromatin pattern with inconspicuous nucleoli (H&E, 40 x). D. Synaptophysin and chromogranin (not shown) immunostains showed strong and diffuse cytoplasmic positivity. Acinar architecture can also be appreciated (synaptophysin, 10 x).")
Case 1. Pathology. A. The cut surface of the tumor displayed well-defined, irregular borders. The variegated lesion was red to yellow, solid and cystic. B. Tumor cells are seen on the left arranged in trabeculae and nests. Residual benign hepatic parenchyma is seen on the right (H&E, 10x). C. On higher power, the tumor cells display round nuclei and moderate amounts of eosinophilic cytoplasm. Nuclei show a characteristic stippled (“salt and pepper”) chromatin pattern with inconspicuous nucleoli (H&E, 40 x). D. Synaptophysin and chromogranin (not shown) immunostains showed strong and diffuse cytoplasmic positivity. Acinar architecture can also be appreciated (synaptophysin, 10 x).
Given that this patient's PHNET is WHO grade 2, both a follow up abdominal MRI and Octreotide scan were performed three months postoperatively, both of which showed no evidence of any recurrence or presence of a previously undiagnosed primary tumor. He also underwent both upper and lower endoscopy which were unremarkable and showed only normal mucosa. EUS was not performed. The patient was most recently seen in clinic nine months postoperatively, and MRI of the abdomen performed at that time also showed no evidence of recurrent disease.
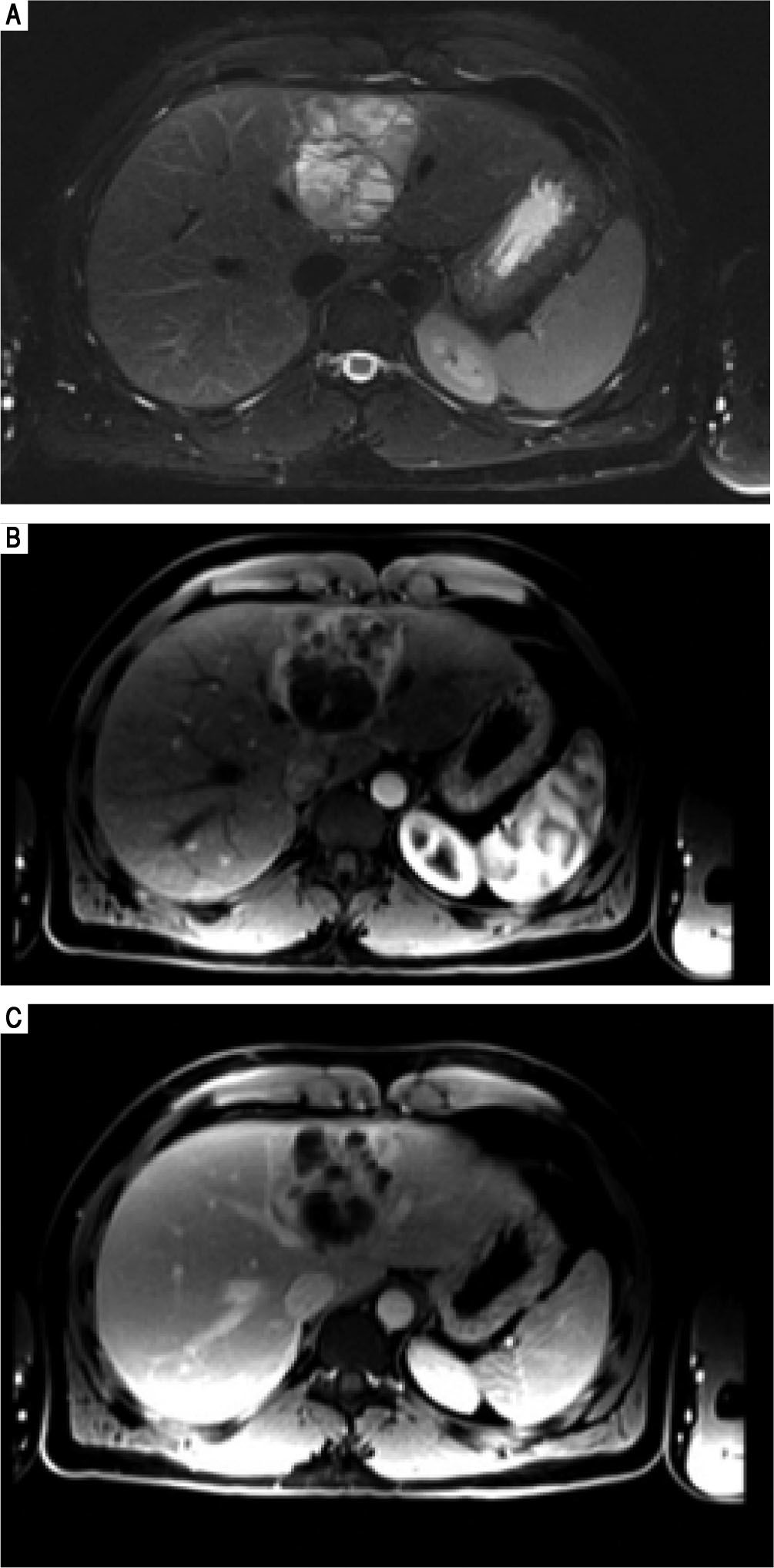
Case 2Case 2 is a 26-year old male with a past medical history significant for obstructive sleep apnea and right knee pain. He was scheduled to undergo right knee surgery for a possible meniscal tear, but during a preoperative transthoracic echocardiogram, a cystic appearing mass was identified within the left lobe of the patient's liver. He underwent follow up abdominal MRI (Figure 3) which demonstrated a 6.8 x 6.5 x 5.8 cm multiseptated mass in the medial segment of the left lobe of the liver. There were no other intra-abdominal lesions noted.
 portal venous phases demonstrates the soft tissue components of the complex cystic mass show arterial phase enhancement (B) and remains slightly hyperintense with respect to hepatic parenchyma on the portal venous phase. This finding can be seen with neuroendocrine tumors, but is not specific.")
Case 2. MRI abdomen. A. Axial fat suppressed T2-weighted MR image demonstrates a complex multiloculated cystic mass originating in the medial segment of the left lobe of the liver. Note the fluid-fluid level within a locule. B. Axial fat-suppressed gadolinium-enhanced T1-weighted MR images in the arterial and (C) portal venous phases demonstrates the soft tissue components of the complex cystic mass show arterial phase enhancement (B) and remains slightly hyperintense with respect to hepatic parenchyma on the portal venous phase. This finding can be seen with neuroendocrine tumors, but is not specific.
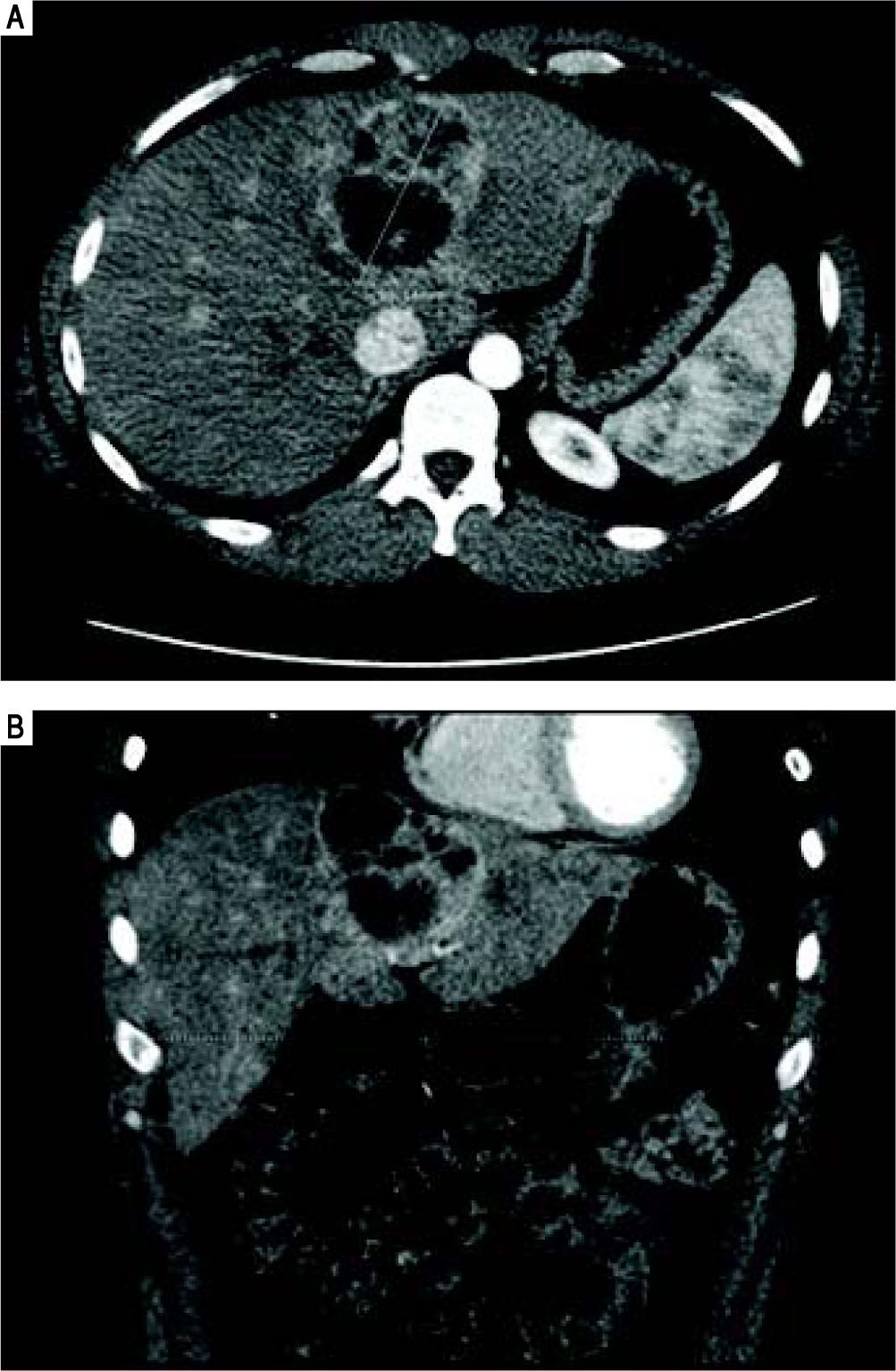
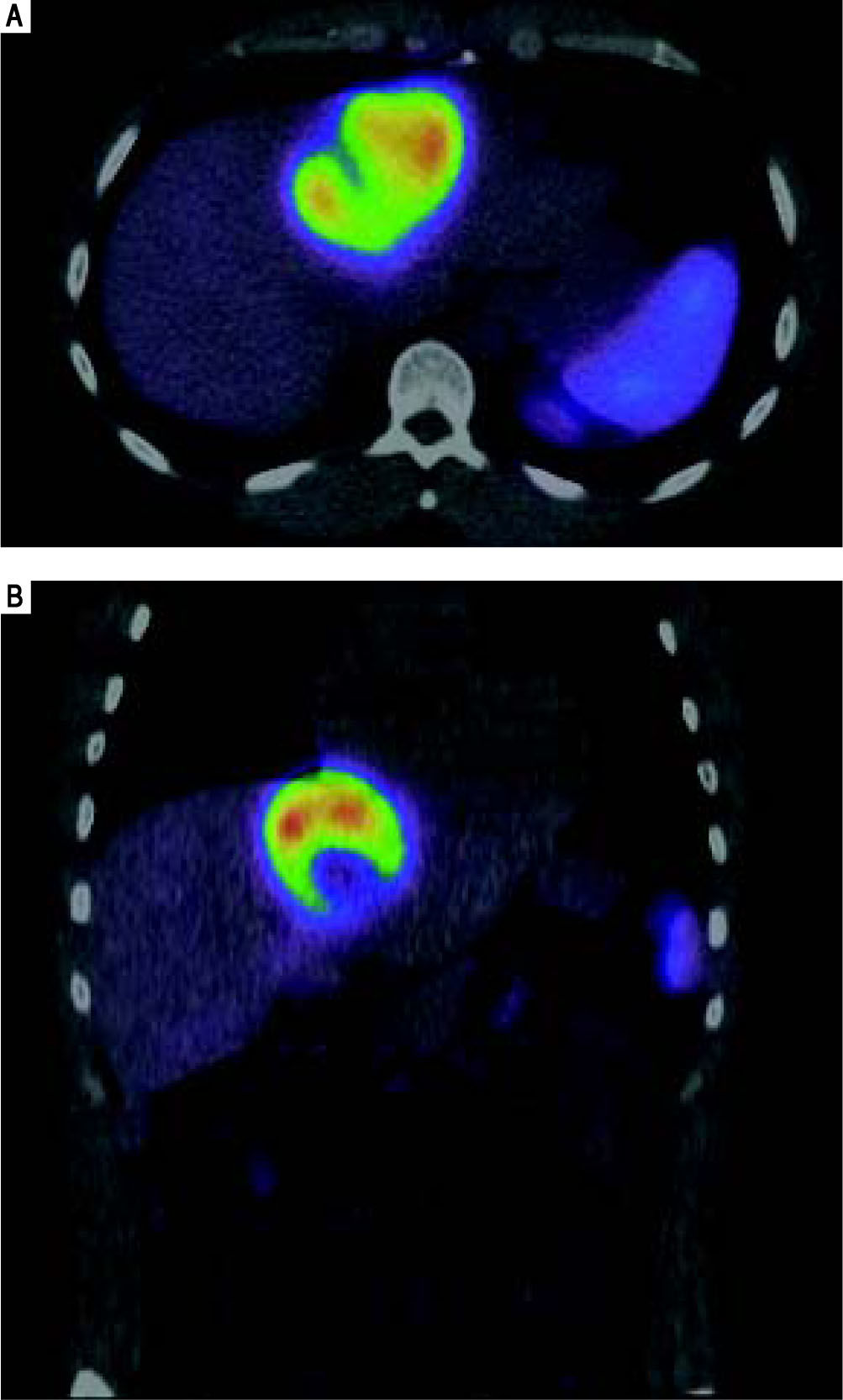
He was referred to GI/Hepatology for further evaluation. During that evaluation, he did report chronic fatigue and occasional mild right-sided abdominal pain, but was otherwise asymptomatic of the lesion. He denied any weight loss, fevers, chills, malaise, flushing, nausea, vomiting, or diarrhea. He had a prior history of overseas travel to Europe and the Middle East with the military, but his last trip was more than 2 years ago. He reported a family history significant for colon cancer and anemia, but no prior history of liver disease or tumors. He is a former smoker, and reported rare social ETOH use. His laboratory profile revealed normal liver function tests (Tbil 0.22, Dbil 0.11, AP 72, AST 26, ALT 24, Alb 4.2), serotonin level 105 (ref.: 56-244 ng/mL), CA 19-9 (6.7, ref.: 0.0 - 37.0 U/mL), and AFP (2, ref.: < 6 ng/mL). His chromogranin A level, however, was found to be elevated at 430 (ref.: 0-95 ng/mL). He underwent a follow up CT chest/abdomen/pelvis at this time (Figure 4), which showed an increase in the size of the left multiloculated cystic hepatic lesion with a maximal diameter now 8.5 cm. No other lesion was identified within the chest or abdomen that could represent a primary lesion. The possibility that the mass represented a neu-roendocrine tumor was made, and the patient was referred for an IN-111 octreotide scan (Figure 5), which showed that the mass demonstrated intense IN-111-octreotide uptake, compatible with a neuroendocrine tumor. There were no areas of additional uptake within the abdomen or pelvis.
 and coronal (B) planes.")
 and coronal (B) planes demonstrates intense activity within the liver mass consistent with a neuroendocrine tumor.")
After diagnosis of a primary non-functioning hepatic neuroendocrine tumor was made, the patient was referred for surgical resection of the mass. He underwent an uncomplicated caudate sparing left liver lobectomy. During the surgery, detailed exploration of the abdominal cavity was negative for any other masses. His postoperative course was unremarkable; he progressed appropriately, and was discharged to home on postoperative day 4. Final surgical pathology (Figure 6) showed a well differentiated neuroendocrine tumor measuring 6.7 cm in maximal diameter, with a mitotic count of 1 per 10 per powered fields, and Ki-67 index < 1%, categorizing it as a low grade tumor (WHO grade 1). The surgical margins were negative and no lymphovascular invasion was identified. Immunostains showed positivity for synaptophysin, chromogranin A, keratin AE1/AE3, and CK7. Whether the tumor was primary vs. metastatic could not be determined from histologic evaluation; however, no other primary sites have been identified, suggesting that this is in fact a primary lesion.
. B. Higher power reveals epithelioid tumor cells with stippled chromatin in a fine vascular network (H&E, 40 x). C. Tumor is diffusely and strongly positive for cytokeratin immunostain AE1/AE3 (AE1/AE3, 10 x). D. Chromogranin and synaptophysin (not shown) displayed strong and diffuse cytoplasmic staining (chromogranin A, 10 x).")
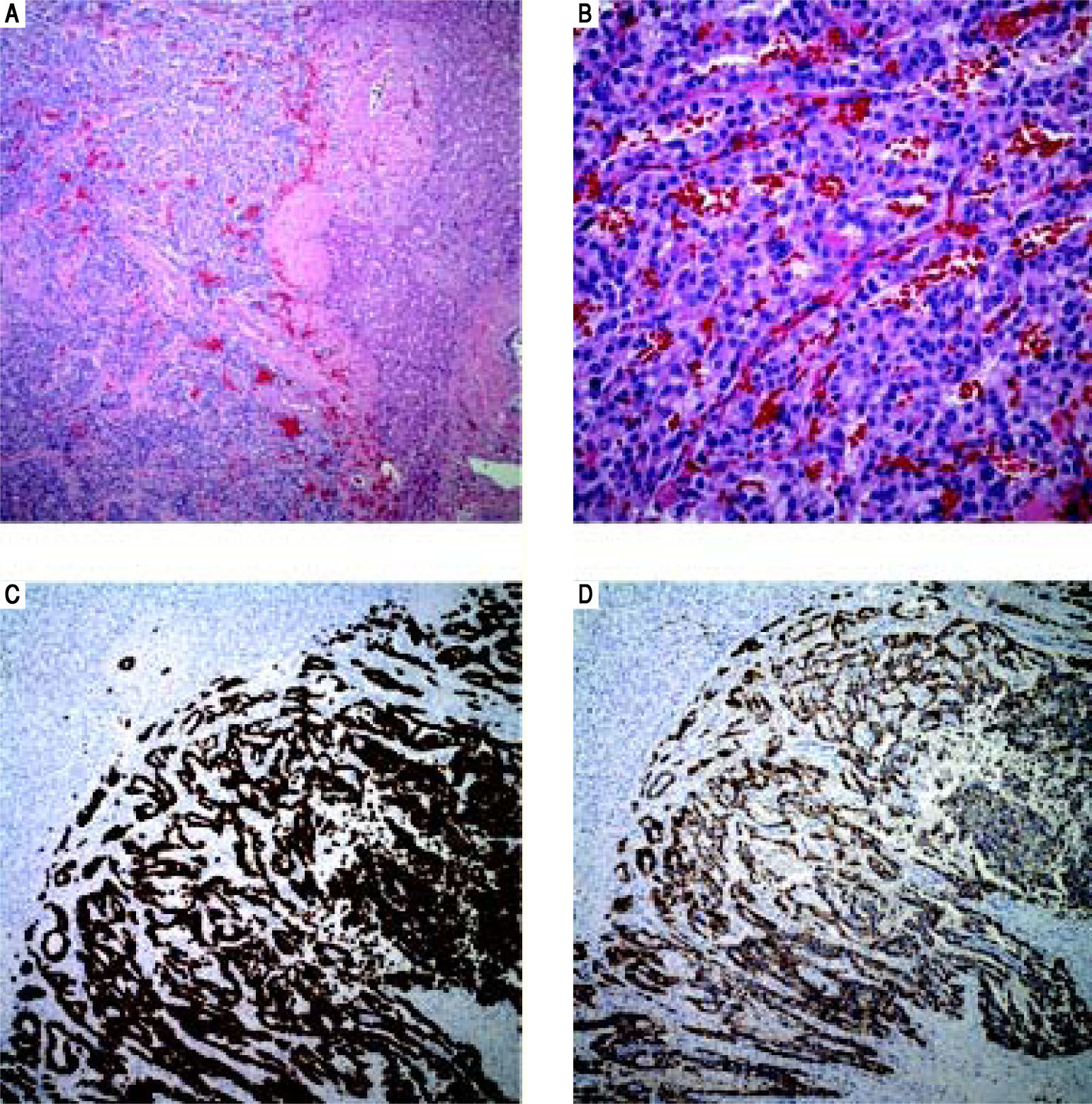
Case 2. Pathology. A. The tumor is arranged in trabeculae and nests with residual hepatic parenchyma seen on the right (H&E, 10x). B. Higher power reveals epithelioid tumor cells with stippled chromatin in a fine vascular network (H&E, 40 x). C. Tumor is diffusely and strongly positive for cytokeratin immunostain AE1/AE3 (AE1/AE3, 10 x). D. Chromogranin and synaptophysin (not shown) displayed strong and diffuse cytoplasmic staining (chromogranin A, 10 x).
Follow up CT scans of the abdomen and pelvis were performed a 6 weeks and 5 months postoperatively, and both were negative for evidence of recurrent disease or other sites of a possible primary lesion. He did not undergo any postoperative upper or lower endoscopy, or EUS. The patient was seen most recently in clinic nine months postoperatively, and an MRI of the abdomen at that time again showed no evidence of recurrent disease. He has not yet had a follow-up Octreotide scan has his tumor is low-grade (WHO grade 1), but this is planned to be performed at the one-year follow-up.
DiscussionWhile NET metastasis to the liver is common, with some studies quoting > 80% of all patients diagnosed with NETs having liver metastasis at the time of diagnosis,4 PHNETs are a rare entity, with fewer than 150 cases reported in the English literature.5 NETs arise from neuroecto-dermal cells, which migrate from the neural crest throughout the body during embryogenesis. These cells do not routinely migrate to the liver, however, which explains why PHNETs are so rare.6 Since these precursor cells are not native to the liver, there are a number of theories to explain the pathogenesis of PHNETs. Hsueh, et al. proposed the presence of ectopic adrenal or pancreatic tissue as the source for PHNET development.7 Alternatively, Alpert, et al. have suggested that argentaffin cells located within the bile duct epithelium are the source of these tumors.8 In both these cases, chronic inflammation of the biliary system could initiate intestinal metaplasia, in turn predisposing to the development of NETs.6,9 A third theory suggests that they arise from neuroendocrine differentiation of a malignant stem cell. Despite the various theories that exist, there have been no studies published specifically defining the pathogenesis of the PHNET disease entity.
Prior systematic reviews of existing case reports have shown that PHNETs are more common in women and most frequently occur in the middle-aged population (4th-5th decades).11-13 They are usually non-functional, and less than 20% of patients present with the flushing, diarrhea, and abdominal pain associated with classic carcinoid syndrome.14 The most common presentations are secondary to mass effect, and include abdominal pain, jaundice, and a palpable mass, followed by an asymptomatic presentation with the tumor identified incidentally,3,9 as was the case with both of our patients. Once the presence of the tumor has been established, it must first be differentiated from other more common hepatic neoplasms, such as hepatocellular carcinoma (HCC), cholangiocarcinoma, metastatic disease from distant primary neoplasms, or ecchinococcal cysts.14
Radiographically, ultrasound, CT, and MRI, have low specificity in differentiating PHNETs from other hepatic tumors, which may be cystic or enhance in the arterial phase.11,15 While there are very few reports of PHNETs in the literature, there are some radiographic findings which may be useful in suggesting the proper diagnosis. Similar to metastatic NET tumors to the liver, PHNETs tend to be hypervascular tumors which markedly enhance,15 and while they are usually solid, cystic PHNETs have been described.16 Both cases presented in this manuscript were primarily cystic in nature and also contained fluid-fluid levels. When a PHNET is suspected, somatostatin receptor scintigraphy (OctreoScan) is performed, using IN-111 labeled octreotide. It is up to 90% sensitive and 83% specific with a 100% positive predictive value for identifying NETs, and has been shown to identify an additional 16% of distant lesions missed by CT and/or MRI.11,17
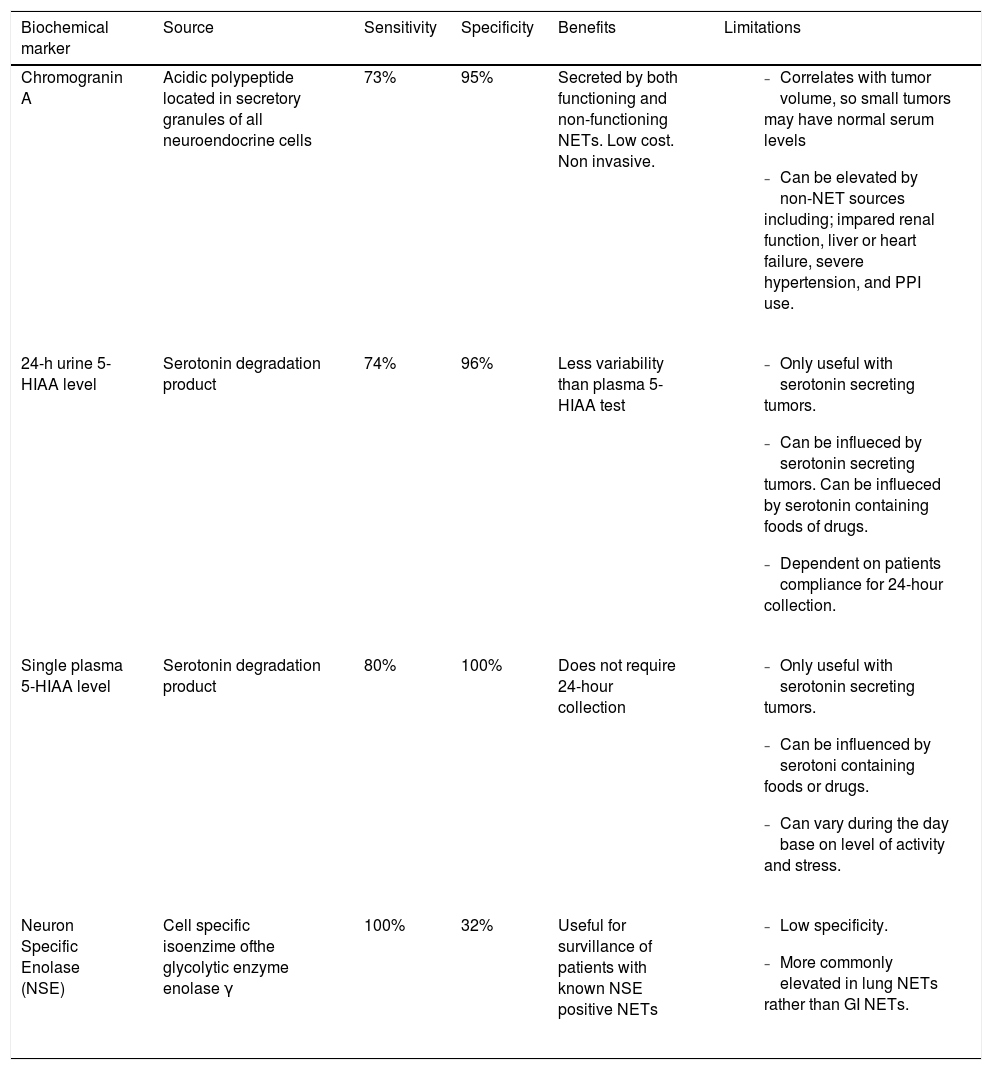
When evaluating hypervascular liver masses, other patient factors must be taken into account to help in differentiating these masses from other hepatic tumors. Patients with HCC often have a history of alcoholic or viral hepatitis, with or without cirrhosis, and elevated serum AFP levels. In contrast, patients with PHNETs do not usually have any of these clinical findings.15 In addition to lacking AFP elevations, these patients also most often have normal CEA and CA 19-9 levels, making these lab tests of little diagnostic value. There are several biochemical markers, however, that have been identified which are useful in helping to diagnose NETs, which include Chromogranin A, 5-Hydroxyindole Acetic Acid (5-HIAA), and Neuron Specific Enolase (NSE) (Table 1).4,18-23
Biochemical markers for NETs.
| Biochemical marker | Source | Sensitivity | Specificity | Benefits | Limitations |
|---|---|---|---|---|---|
| Chromogranin A | Acidic polypeptide located in secretory granules of all neuroendocrine cells | 73% | 95% | Secreted by both functioning and non-functioning NETs. Low cost. Non invasive. |
|
| 24-h urine 5-HIAA level | Serotonin degradation product | 74% | 96% | Less variability than plasma 5-HIAA test |
|
| Single plasma 5-HIAA level | Serotonin degradation product | 80% | 100% | Does not require 24-hour collection |
|
| Neuron Specific Enolase (NSE) | Cell specific isoenzime ofthe glycolytic enzyme enolase γ | 100% | 32% | Useful for survillance of patients with known NSE positive NETs |
|
5-HIAA = 5-Hydroxyindole Acetic Acid.
While imaging and laboratory evaluation can suggest the diagnosis of a NET over other more common liver tumors, the only means of definitive diagnosis is by patho-logic evaluation of a surgically resected specimen. Needle biopsy is not recommended as it has been shown to have a variable diagnostic accuracy of only 11-50%, and it does in-crease the risk of tumor seeding.3,24 If there is a high index of suspicion for a NET, a thorough survey of the abdominal cavity should be made at the time of resection to evaluate for foci of distant disease. As such, a diagnosis of a PHNET cannot be confidently made without a combination of a NET diagnosis on pathologic evaluation, a negative abdominal survey at the time of resection, and a negative work up for extrahepatic disease after all imaging studies have been completed in both the pre- and postoperative settings.24
Well differentiated neuroendocrine tumors (WD-NETs) show characteristic histomorphology with nested, trabecular and/or acinar architectural patterns. The epithelioid tumor cells have moderate amounts of eosinophilic cytoplasm and round nuclei with a granular/stippled chromatin pattern, which is often referred to as “salt and pep-per”. While the morphology is often strongly suggestive of a neuroendocrine tumor, it should be noted that hepatocellular carcinoma can have a similar growth pattern (trabecular and/or acinar) and also displays cells with moderate amounts of eosinophilic cytoplasm. Hepatocellular carcinoma may show bile production however, a feature that is not seen in WDNETs. Hepatocyte specific markers, such as HepPar1 and arginase 1 further aid in this distinction and are generally negative in WDNETs and positive in most hepatocellular carcinomas. Further confirmation of the diagnosis of PHNET can be made with immunohistochemical stains, which show positivity for synaptophysin, chromogranin, CD56 (NCAM), and cytokeratins in the majority of cases. This morphologic and immunohistochemical pattern are common across WD-NETs of any site and, while some additional tests may suggest a possible primary site (e.g., CDX2 positivity may suggest gastrointestinal origin), determining whether a WDNET represents a primary or metastatic focus relies highly on the clinical context. The vast majority of WD-NETs involving the liver are metastatic in nature, generally from small intestinal or pancreatic WDNETs.
WDNETs are graded based on mitotic rate and Ki-67 proliferation index, independent of primary location. Ki-67 is a known marker of tumor proliferation, and elevated indices have been associated with worse prognosis in gastrointestinal NETs.24 In 2010, the WHO reorganized its categorization of NETs into low and intermediate grade neuroendocrine neoplasms (grade 1 and 2), and high grade neuroendocrine carcinomas (grade 3), the latter of which is being increasingly recognized as a distinct entity from a standard WDNET.25,26 These grade 3 tumors are often characterized by conspicuous mitotic activity (usually 40-50 mitoses per 10 high power fields and/or a Ki-67 index > 20%) and high grade cytologic features, which may show the classic morphology of small cell carcinoma or large cell neuroendocrine carcinoma. While this grading system is designed for NETs in the more common locations of small bowel, stomach, colon, and ampulla of Vater, it has been adopted to the description of PHNETs as well. The tumor grade on pathologic evaluation is important for prognosis, specifically the Ki-67 index. Patients with malignant pancreatic NETs and a Ki-67 index < 2% have been shown to have a better prognosis than those with an index ≥ 2%. This has been shown to hold true for patients with PHNETs, where the average Ki-67 index in patients without recurrent disease was found to be 1.7%.24
Surgery with complete resection and negative margins is widely understood to be the treatment modality of choice for PHNETs, as up to 85% of tumors are resectable,12 and surgery yields a very good long term 5-year survival at 74-78%.3,6,13,24,27-29 The extent of hepatic resection does not seem to impact survival, as survival rates do not differ in patients receiving simple wedge resections compared to those receiving anatomic resections (segmentectomies or lobectomies) for similar tumors.27 Despite appropriate surgical resection, recurrence rates remain as high as 19.8%.3 Other treatment modalities aside from surgery exist, but they have been less well studied, and thus, their role in treatment of these tumors is less well understood. Liver transplantation or transarterial chemoembolization (TACE) have also been suggested as alternative treatment modalities in patients with surgically unresectable disease. TACE has the added benefit of being cytoreductive, and can be used to downstage previously unresectable tumors to a size and distribution where resection becomes possible.3,28,30 Chemotherapy has a limited role in the treatment of these tumors, and its role is confined to patients with unresectable primary disease, or those patients with distant metastases.3 There has been no evidence to suggest a survival benefit to receiving preoperative chemotherapy, radiation therapy, or TACE in patients with resectable disease.13
A question often raised is whether or not to perform a prophylactic appendectomy at the time of liver resection? This practice is not currently supported by the literature, even if there is suspicion that the liver lesion is in fact a metastasis from an unidentified primary NET. To the contrary, several recent studies have suggested that appendiceal NETs are very unlikely to metastasize to the liver. Geramizadeh, et al. recently showed that of 20 patients with primary appendiceal NET, none showed metastasis to the liver.5 Similarly, in a large database study out of Sweden by Riihimaki, et al. using the Swedish Cancer Registry, the odds ratio of primary appendiceal NET metastasizing to the liver was 0.02.4 These findings would suggest that there is little benefit to performing a prophylactic appendectomy at the time of liver resection.
ConclusionPHNETs are rare but distinct pathologic entities which, when treated appropriately, have a very good long term prognosis. Surgical resection is the mainstay of therapy, and is the only treatment modality that has been shown to render a good long term survival. Chemotherapy, radiation therapy, or TACE can be used in patients with unresectable disease for down staging or in preparation for liver transplantation. Given that the recurrence rate is high despite complete surgical resection (19.8%), close follow up is of paramount importance. There are no currently established protocols delineating the role of adjunct therapies in preventing post-resection recurrence for PHNET.



