





La disbetalipoproteinemia o hiperlipoproteinemia tipo III es una hiperlipemia mixta grave consecuencia de la acumulación de partículas remanentes de quilomicrones y VLDL en plasma, también llamadas β-VLDL. Se produce por un defecto en el reconocimiento por parte de los receptores hepáticos LDL y LRP de las β-VLDL. Mutaciones en el gen de APOE, especialmente los sujetos homocigotos para el alelo ɛ2/ɛ2, son las responsables de esta falta de reconocimiento por los receptores.
La disbetalipoproteinemia representa el 2-5% de las dislipemias mixtas atendidas en las unidades de lípidos; es altamente aterogénica y predispone a una ateromatosis difusa, tanto coronaria, vascular periférica y carotídea, por lo que es necesario un diagnóstico y tratamiento precoz. La presencia de hipertrigliceridemia, con cocientes colesterol no-HDL/apolipoproteína B>1,43 (en mg/dL), seguido de genotipado de APOE es el método de elección en el diagnóstico de la disbetalipoproteinemia. Es una dislipemia que responde bien al tratamiento higiénico-dietético, aunque muchas veces la combinación estatina y fenofibrato es necesaria para lograr un control óptimo.
Dysbetalipoproteinaemia (or type III hyperlipoproteinaemia) is a severe mixed hyperlipidaemia resulting from the accumulation of remnant chylomicron and VLDL particles in plasma, also called β-VLDL. It is caused by a defect in the recognition by hepatic LDL and lipoprotein receptor-related protein (LRP) of β-VLDL. Mutations in the APOE gene, especially in subjects homozygous for the ɛ2/ɛ2 allele, are responsible for this lack of receptor recognition.
Dysbetalipoproteinaemia represents 2-5% of the mixed dyslipidaemias seen in Lipid Units, is highly atherogenic and predisposes to diffuse atheromatosis, either coronary, peripheral vascular, or carotid, so early diagnosis and treatment is necessary. The presence of hypertriglyceridaemia, with non-HDL cholesterol/apolipoprotein B ratios>1.43 (in mg/dL) followed by APOE genotyping is the method of choice in the diagnosis of dysbetalipoproteinaemia. It is a dyslipidaemia that responds well to hygienic-dietary treatment, although the combination of statin and fenofibrate is often necessary to achieve optimal control.
La disbetalipoproteinemia (DBLP), o hiperlipoproteinemia tipo III (HLPIII) en algunas situaciones, se caracteriza por la acumulación en plasma de partículas remanentes de las lipoproteínas ricas en triglicéridos (TG), es decir, de las partículas resultado del catabolismo periférico de las lipoproteínas de muy baja densidad (VLDL) y quilomicrones (QM). La acumulación de estos remanentes se produce por un trastorno en su catabolismo hepático consecuencia de variantes genéticas disfuncionales o ausencia de la apolipoproteína E (apo E). Desde el punto de vista conceptual, la DBLP se define por el aumento de remanentes con cocientes de colesterol en VLDL/TG>0,3; y la HLPIII cuando coexiste DBLP con concentraciones elevadas de colesterol y TG. Por tanto, toda HLPIII tiene DBLP, pero lo contrario no es cierto. Muchos sujetos con el genotipo ɛ2/ɛ2 de APOE tienen DBLP, pero solo unos pocos llegan a desarrollar una auténtica HLPIII. Sin embargo, esta distinción conceptual no se suele aplicar con frecuencia y en la práctica DBLP, DBLP familiar (DF) e HLPIII suelen hacerse sinónimos. Nosotros en esta revisión utilizaremos indistintamente dichos términos al referirnos a la HLPIII.
La DBLP fue descrita por McGinley et al. en 1952 durante la caracterización por ultracentrifugación de un grupo de 14 sujetos con diferentes tipos de xantomas1. No fue hasta 1967 cuando tuvo una descripción clínica y bioquímica más detallada y fue denominada HLPIII en la famosa clasificación de las hiperlipoproteinemias de Fredrickson et al.2.
EpidemiologíaLa HPLPIII es una dislipemia rara, probablemente solo alrededor de 1 cada 5.000-10.000 sujetos en la población general la padecen, aunque las estimaciones varían notablemente entre estudios. La frecuencia del genotipo ɛ2/ɛ2 de APOE es de aproximadamente el 1% en la población en general, por lo que solamente una pequeña proporción de los sujetos con la predisposición genética llegarían a desarrollar una verdadera DF3. Sin embargo, en un análisis transversal en 5.272 participantes de la Encuesta Nacional de Examen de Salud y Nutrición de los EE.UU. (NHANES) entre 2011 y 2014 y en 128.506 participantes del estudio VLDL (Very Large Database of Lipids Study) la prevalencia en la población fue en torno al 2%, con una prevalencia creciente con la edad, pasando del 1,6% antes de los 40 años al 2,8% en personas ≥75años, sin diferencias entre hombres y mujeres. La diabetes y la obesidad fueron los factores más frecuentemente asociados a la presencia de DBLP4. En este estudio, la DBLP se definió únicamente en base a las concentraciones de colesterol total, TG y apolipoproteína B (apo B<120mg/dL, TG>133mg/dL, TG/apo B<8,8 y colesterol/apo B<2,4), sin genotipado de APOE. Estos criterios son sensibles, pero poco específicos, por lo que la prevalencia posiblemente se sitúe en una cifra intermedia. En nuestra unidad, la frecuencia de DF definida por criterios genéticos y lipídicos es del 3,7% (34/915 sujetos) entre aquellos con dislipemia mixta (Bea et al., en preparación). Es decir, en nuestra unidad de lípidos, aproximadamente uno de cada 30 sujetos con hiperlipemia mixta tendría una DBLP. El registro de dislipemias de la Sociedad Española de Arteriosclerosis sería una fuente excelente para validar estas cifras5.
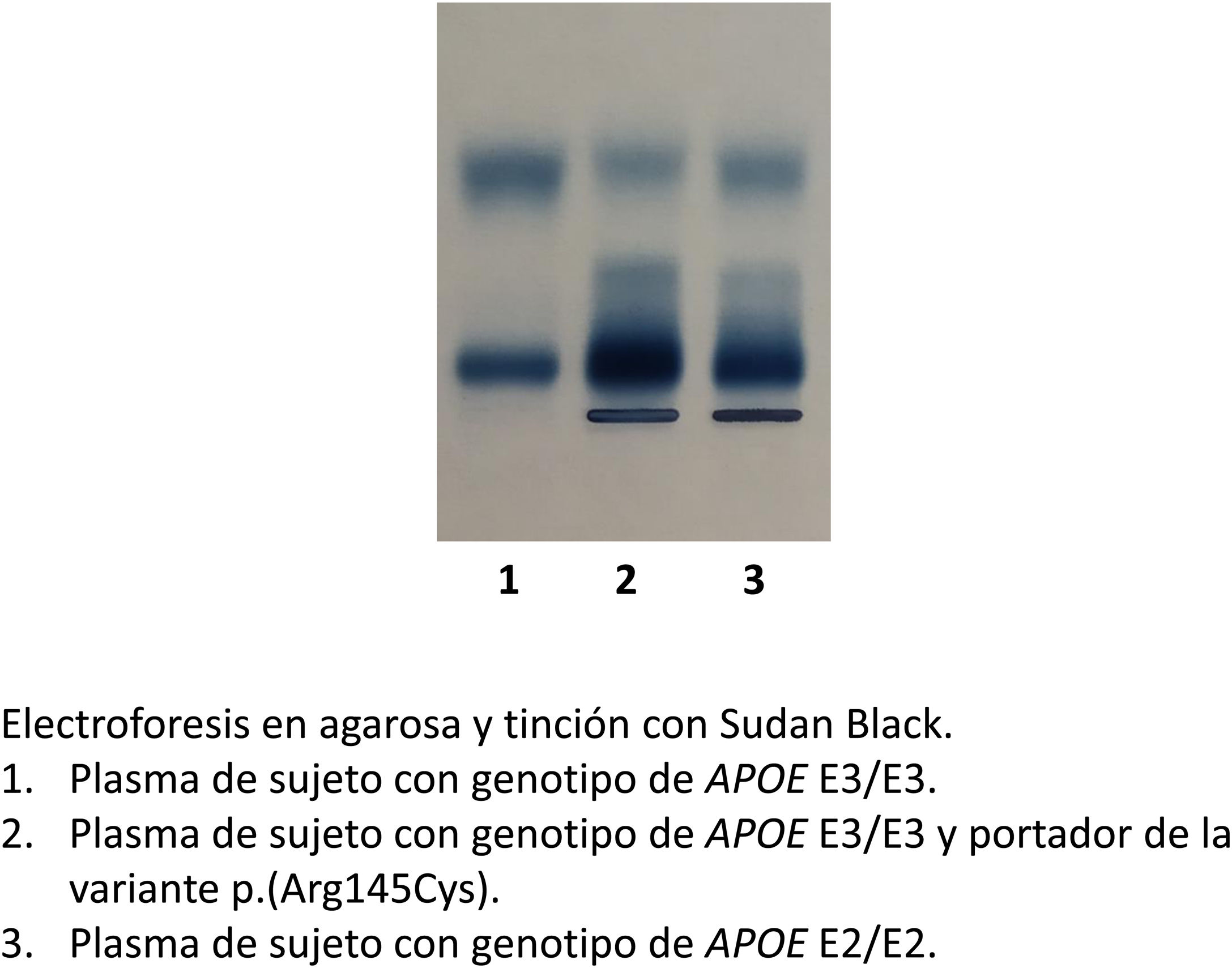
EtiopatogeniaLa DBLP se caracteriza por una hiperlipemia mixta debida a la acumulación de lipoproteínas remanentes de origen hepático e intestinal, es decir, VLDL y QM remanentes que dan lugar en una electroforesis a una banda β ancha, o β-VLDL, y que producen hipercolesterolemia e hipertrigliceridemia, con elevaciones aproximadamente equimolares de colesterol y TG6. En situación normal, los QM y VLDL remanentes se retiran rápidamente de la circulación plasmática por endocitosis mediada por receptor en el hígado, mediante la unión de la apo E presente en estas lipoproteínas al receptor de las lipoproteínas de baja densidad (LDL) (rLDL) y a la proteína relacionada con el rLDL (LRP). Sin embargo, en la DBLP, el defecto en la retirada de las partículas remanentes se debe a un defecto en apo E, de manera que no se une eficientemente a sus receptores. El defecto en el procesado de estas lipoproteínas ricas en TG tiene varias consecuencias: 1) Debido a que su tiempo de residencia en el plasma es más prolongado de lo normal, estas partículas se acumulan en gran número en el plasma. 2) Durante este prolongado período en la circulación, las partículas se convierten en ricas en colesterol, ya que adquieren un exceso de ésteres de colesterol debido al intercambio lipídico mediado por la proteína transferidora de ésteres de colesterol (CETP). Como consecuencia, estas partículas remanentes anormales tienen elevados los cocientes colesterol/triglicéridos y colesterol/apo B, así como unas propiedades electroforéticas anormales. 3) Debido a que las VLDL no se convierten en LDL en la proporción adecuada, el número de partículas LDL es bajo en relación con partículas VLDL, siendo la concentración total plasmática de apo B normal. De acuerdo con esto, los cocientes de apo B VLDL/apo B LDL y apo B VLDL/apo B total son más elevados de lo normal7,8. Así, la DBLP es una excepción a la regla comúnmente aceptada de que el riesgo cardiovascular relacionado con lipoproteínas está directamente relacionado con la cantidad de apo B plasmática.
Apo E y sus isoformasLa apo E es una proteína rica en arginina descubierta en los años 70 en las lipoproteínas ricas en triglicéridos, siendo en la actualidad una de las más estudiadas, tanto por su papel fundamental en el metabolismo lipoproteico y en la enfermedad cardiovascular como por sus funciones neurológicas en la enfermedad de Alzheimer. Se sintetiza como un prepéptido de 317 aminoácidos, y tras una escisión post-traduccional de un péptido señal de 18 aminoácidos, resulta en una proteína madura de 299 aminoácidos. La apo E se sintetiza y secreta por muchos tejidos, principalmente el hígado, el cerebro, la piel y los macrófagos tisulares de todo el organismo, sitios donde la apo E desempeña funciones importantes9. El gen que la codifica es APOE, localizado en el cromosoma 19, y formado por 3.597 nucleótidos, repartidos en 4 exones y 3 intrones.
En humanos, existen 3 isoformas comunes de apo E, llamadas apo E2, apo E3 y apo E4, de acuerdo con su movilidad en una electroforesis de isoelectroenfoque. Cada una de estas isoformas difiere en su punto isoeléctrico en una unidad de carga, siendo apo E4 la isoforma más básica y apo E2 la más ácida. La biosíntesis de estas isoformas está bajo el control de 3 alelos independientes. La isoforma apo E3 es la más frecuente en la población, y tiene una cisteína en posición 112 (Cys112) y una arginina en posición 158 (Arg158). Las isoformas apo E2 y apo E4 difieren de apo E3 por una sustitución de un aminoácido en una única posición: apo E2 tiene Cys112 y Cys158 y apo E4 tiene Arg112 y Arg158. Estas 3 isoformas comunes, apo E2, apo E3, y apo E4, ocurren con una frecuencia alélica de aproximadamente 0,10, 0,75 y 0,15, respectivamente, en la mayoría de las poblaciones caucásicas10.
Las variantes génicas que dan lugar a las isoformas de apo E son los SNV (single nucleotide variant) rs429358 para el codón 112 y el SNV rs7412 para el codón 158. Existen 6 posibles combinaciones de estos SNV, de manera que hay 6 posibles genotipos frecuentes: ɛ2/ɛ2, ɛ2/ɛ3, ɛ2/ɛ4, ɛ3/ɛ3, ɛ3/ɛ4, y ɛ4/ɛ4, con una prevalencia aproximada de 1, 22, 2, 58, 14 y 3%, respectivamente, en poblaciones caucásicas.
Las isoformas comunes de apo E contribuyen a la variabilidad en los niveles de colesterol plasmático en la población general. Los sujetos con el alelo ɛ2 tienen altos niveles plasmáticos de apo E y bajos niveles de colesterol total, colesterol-LDL y apo B, mientras que los sujetos portadores del alelo ɛ4 presentan el fenotipo opuesto11.
La mayoría de los pacientes con DBLP son homocigotos para la isoforma E2 de apo E12, que presenta una unión defectuosa a los receptores hepáticos de lipoproteínas, y por tanto, se produce una retirada retardada del plasma13. Sin embargo, se necesitan otros factores adicionales genéticos o ambientales para que se desarrolle la enfermedad, ya que solo el 1-4% de los homocigotos ɛ2/ɛ2 desarrollan DBLP. Por tanto, la mayoría de homocigotos ɛ2/ɛ2 son normolipémicos o incluso hipolipémicos, pero con β-VLDL en su plasma, por lo que deberían clasificarse también como DBLP sin HLPIII. Una minoría, menos del 10%, de los pacientes con HLPIII son portadores heterocigotos de variantes genéticas de APOE con un modelo de herencia dominante. La mayoría de estas variantes resultan de sustituciones un único aminoácido: p.(Arg136Ser), p.(Arg136Cys), p.(Arg142Cys), p.(Arg142Leu), p.(Arg145Cys), p.(Arg147Trp), p.(Lys146Asn), p.(Lys146Gln) y p.(Lys146Glu)14.
En 1996, nuestro grupo de investigación publicó la presencia de la mutación p.(Arg136Ser) en familias con HLPIII, demostrando un patrón de herencia de dominancia incompleta15,16. Más recientemente, en 2012, demostramos que casi el 2% de los sujetos con un fenotipo de hiperlipemia familiar combinada eran portadores de esta variante p.(Arg136Ser), por lo que podemos concluir que esta mutación en APOE tiene una frecuencia elevada en población española, siendo una causa frecuente de hiperlipoproteinemia tipo III con herencia dominante en nuestra población17.
Mecanismo de la alteración con receptoresLa apo E normal tiene una alta afinidad por el rLDL posiblemente porque en las lipoproteínas ricas en TG que contienen varias moléculas de apo E por partícula, una vez delipidadas, el dominio de apo E ligando del rLDL se expone con facilidad18. El dominio de apo E ligando del rLDL comprende los aminoácidos 136-150 y es rico en residuos de arginina y lisina cargados positivamente. En ausencia de la Arg158, es decir, en la isoforma apo E2, se posibilita que el Asp154 forme un puente con la Arg150, lo que limita la unión al rLDL pero no al LRP y ni a los proteoglicanos del endotelio hepático, y por tanto, solo una proporción de homocigotos ɛ2/ɛ2 desarrollaría DBLP19. Mutaciones dentro del dominio de unión al rLDL, como la p.(Arg136Ser), modifican la unión a todos los receptores, y de ahí que un único alelo sea capaz de provocar la dislipemia16.
Manifestaciones clínicasLa DBLP se presenta como una enfermedad autosómica recesiva en la mayor parte de los casos y suele comenzar en edades medias de la vida. Tradicionalmente se ha considerado algo más frecuente en varones y suele acompañarse de diferentes condiciones que aumentan la síntesis de partículas VLDL, como la obesidad, el síndrome metabólico o la diabetes mellitus. La DBLP presenta 3 manifestaciones importantes: dislipemia mixta, xantomas cutáneos y ateromatosis generalizada.
Dislipemia mixtaEl patrón lipoproteico es variado y no todos los pacientes tienen el mismo fenotipo. El colesterol y los triglicéridos plasmáticos están elevados con cifras en torno a 300-800mg/dL cada uno y de forma relativamente constante las cifras de ambos son bastante homogéneas, ya que los remanentes acumulados en sangre transportan cantidades semejantes de colesterol y TG. Sin embargo, en presencia de obesidad severa o diabetes descompensada, la concentración de TG puede incluso doblar a la concentración de colesterol. Una característica distintiva es la presencia de lipoproteínas anormales denominadas β-VLDL, debido a su migración β-electroforética en lugar de la migración pre-β habitual de las VLDL típicas (fig. 1). Las β-VLDL son restos enriquecidos en colesterol de origen intestinal y hepático que también están enriquecidos en apo E. La apo B no suele estar elevada y llama la atención su concentración normal-baja a pesar de concentraciones elevadas de colesterol total. El colesterol-HDL y la apolipoproteína A1 están reducidos como ocurre en la mayor parte de las hipertrigliceridemias sin tener características especiales, salvo su enriquecimiento proporcional en TG20. El colesterol-LDL es normal o reducido a pesar de la grave hipercolesterolemia, hecho diferencial con otras formas de hiperlipemia mixta como la hiperlipemia familiar combinada.
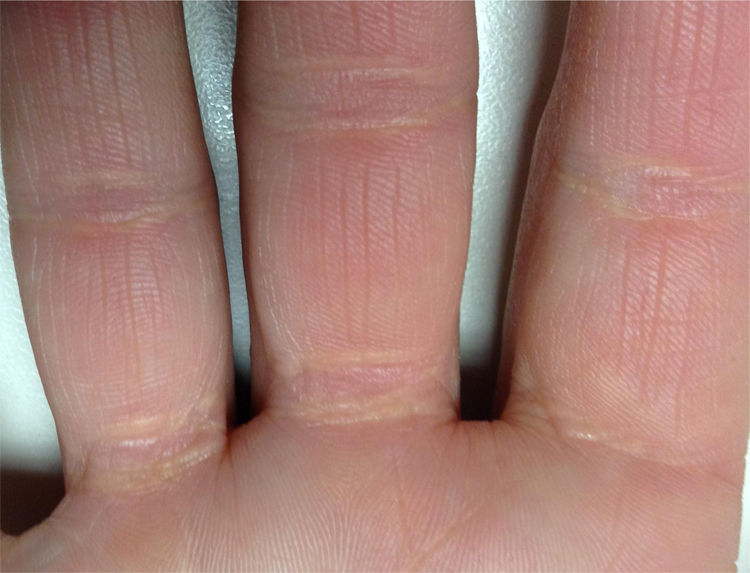
Xantomas cutáneos
Cada día se ven con menos frecuencia, ya que los diferentes tratamientos logran reducirlos o eliminarlos de forma relativamente rápida, y dado su pequeño tamaño y que el uso de hipolipemiantes se ha generalizado, muchas veces pasan desapercibidos para el paciente y para el médico. Sin embargo, son muy específicos, aunque no patognomónicos21. Son de 2 tipos: xantomas planos estriados en las palmas de las manos y pliegues interdigitales de color amarillo anaranjado (fig. 2), y xantomas tubero-eruptivos en codos, rodillas y tendón de Aquiles22.
Enfermedad cardiovascular
El riesgo de enfermedad cardiovascular prematura (CHD) está muy elevado en la DBLP. Hopkins et al., estudiando a 653 sujetos con enfermedad coronaria prematura, identificaron que el 3,4% padecían una DBLP y que el riesgo, independiente de otros factores de riesgo asociados, era entre 5-10 veces superior al de la población general23. En un estudio cooperativo europeo incluyendo 305 pacientes, la prevalencia de enfermedades cardiovasculares fue del 19% para enfermedad coronaria, 11% para enfermedad arterial periférica y 4% para enfermedad cerebrovascular, con un riesgo 10 veces superior de enfermedad cardiovascular prematura en comparación con la población general24. Los factores de riesgo tradicionales también juegan un papel fundamental al explicar el riesgo de estos pacientes25.
DiagnósticoEl diagnóstico convencional de DBLP está basado en la demostración de un aumento en las partículas remanentes anormales. Esto requiere separación de lipoproteínas por ultracentrifugación y, posteriormente, cuantificación de colesterol y triglicéridos o electroforesis del sobrenadante con una densidad<1.006g/mL. El gold standard en los criterios de Fredrickson para el diagnóstico de DBLP incluye: TG entre 150-1.000mg/dL y un cociente colesterol-VLDL/TG>0,30mg/dL. Se han propuesto criterios más simples para el diagnóstico de DBLP que incluyen los cocientes: apo B/colesterol total, colesterol no-HDL/apo B y colesterol remanentes/TG, así como el propuesto por Sniderman y mencionado anteriormente: apo B<120mg/dL, TG>133mg/dL, TG/apo B<8,8 y colesterol/apo B<2,46. En 2 estudios recientes, el cociente colesterol no-HDL/apo B>1,43 en mg/dL seguido de genotipado de APOE mostraron tener la mejor sensibilidad y especificidad en el diagnóstico de DBLP7,8. Debido a la presencia de mutaciones dominantes en APOE diferentes a los SNV comunes, se requiere una secuenciación completa de APOE, o al menos del exón 4, para detectarlas, ya que se han descrito al menos 30 mutaciones diferentes en 20 residuos diferentes de la proteína3.
TratamientoLos pacientes con HLPIII suelen ser muy sensibles al tratamiento higiénico-dietético y en algunos casos, si se corrige la obesidad, los malos hábitos alimenticios o el hipotiroidismo, se normalizan los niveles plasmáticos de lípidos. Por este motivo es imprescindible una dieta hipocalórica en presencia de sobrepeso u obesidad, ausencia de alcohol e hidratos de carbono simples y aumento de la actividad física como primera medida en estos pacientes. En la mayoría de los pacientes con HLPIII, además de la dieta, la medicación hipolipemiante es necesaria para normalizar niveles plasmáticos de colesterol y TG. Uno de los estudios pioneros en este campo fue realizado por nuestro grupo hace más de 20 años26. Comparamos, en un estudio cruzado aleatorizado, doble ciego, controlado con placebo, los efectos de gemfibrozil (1.200mg/día) y simvastatina (20mg/día) sobre lípidos, apo AI, apo B y apo E, y sobre lípidos y contenido de apo B en VLDL, IDL, LDL y HDL en 10 pacientes con DBLP.
Los niveles de colesterol total, colesterol VLDL, colesterol IDL y apo B disminuyeron con ambos fármacos. Mayores reducciones de triglicéridos se obtuvieron con gemfibrozil en comparación con simvastatina. La reducción del colesterol fue más eficaz con simvastatina que con gemfibrozil. Los niveles de IDL y apo E se redujeron de manera similar con ambos fármacos. Sin embargo, ninguno de los 2 fármacos por separado logró un control completo de la dislipemia26. En nuestra experiencia, la combinación de estatinas de mediana potencia junto con fenofibrato es el tratamiento de elección en estos pacientes y logra la normalización lipídica en la mayor parte de los pacientes, reservando las estatinas de alta potencia en casos difíciles de controlar o en prevención secundaria27,28.
Conflicto de interesesLos autores declaran no tener conflictos de interés con el contenido del presente trabajo.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.
Este trabajo ha sido financiado con los proyectos PI18/01777, PI19/00694 y CIBERCV (CB16/11/00451); y Gobierno de Aragón (Grupo Consolidado B-14).