









El acúmulo de quilomicrones en el plasma más allá del período postprandial es un hecho patológico secundario a la falta de actividad parcial o completa de la lipoproteinlipasa que puede conducir a episodios recurrentes de dolor abdominal y pancreatitis aguda. En este artículo, se repasa la fisiopatología de este síndrome y las características diferenciales según se deba a causas monogénicas congénitas o adquiridas sobre una base poligénica en la que pueden intervenir múltiples factores.
The accumulation of chylomicrons in plasma beyond the postprandial period is a pathological event secondary to the partial or complete lack of activity of lipoprotein lipase that can lead to recurrent episodes of abdominal pain and acute pancreatitis. This article reviews the pathophysiology of this syndrome and the differential characteristics depending on whether it is due to congenital monogenic causes or acquired on a polygenic basis in which multiple factors may inluence.
Desde un punto de vista histórico, el síndrome de quilomicronemia (SQM) fue por primera vez descrito en un grupo de pacientes1, la mayoría diabéticos, con marcada hipertrigliceridemia y un cuadro clínico caracterizado por dolor abdominal, pancreatitis, lipemia retinalis, xantomas eruptivos y alteración mental, que aparecía con el consumo de alcohol, incluso en pequeñas cantidades.
Desde un punto de vista biológico, el SQM refleja un hecho patológico, la presencia de quilomicrones (QM) en sangre tras un período de ayuno (≥ ocho horas), detectable mediante ultracentrifugación (técnica engorrosa reservada para investigación) y que puede sospecharse en las mismas condiciones de ayuno por el aspecto «en salsa de tomate» de la sangre recién extraída, la apariencia «lechosa» del suero tras la centrifugación, su «fenotipo» característico posterior al refrigerado a 4°C durante 16 a 18 horas (fig. 1)2 o simplemente por la presencia de hipertrigliceridemia intensa (generalmente > 1.000 mg/dL)3.
 tiene un aspecto pajizo y traslúcido (fenotipo IIa de Fredrickson). En casos de síndrome de quilomicronemia, el suero refrigerado puede aparecer como un sobrenadante lechoso (QM) e infranadante traslúcido o fenotipo I de Fredrickson (centro; quilomicronemia familiar) o como un sobrenadante lechoso (QM) e infranadante turbio (VLDL) o fenotipo V de Fredrickson (derecha; quilomicronemia multifactorial). QM: quilomicrones; VLDL: lipoproteínas de muy baja densidad.")
En casos de hipercolesterolemia pura el suero refrigerado (izquierda) tiene un aspecto pajizo y traslúcido (fenotipo IIa de Fredrickson). En casos de síndrome de quilomicronemia, el suero refrigerado puede aparecer como un sobrenadante lechoso (QM) e infranadante traslúcido o fenotipo I de Fredrickson (centro; quilomicronemia familiar) o como un sobrenadante lechoso (QM) e infranadante turbio (VLDL) o fenotipo V de Fredrickson (derecha; quilomicronemia multifactorial).
QM: quilomicrones; VLDL: lipoproteínas de muy baja densidad.
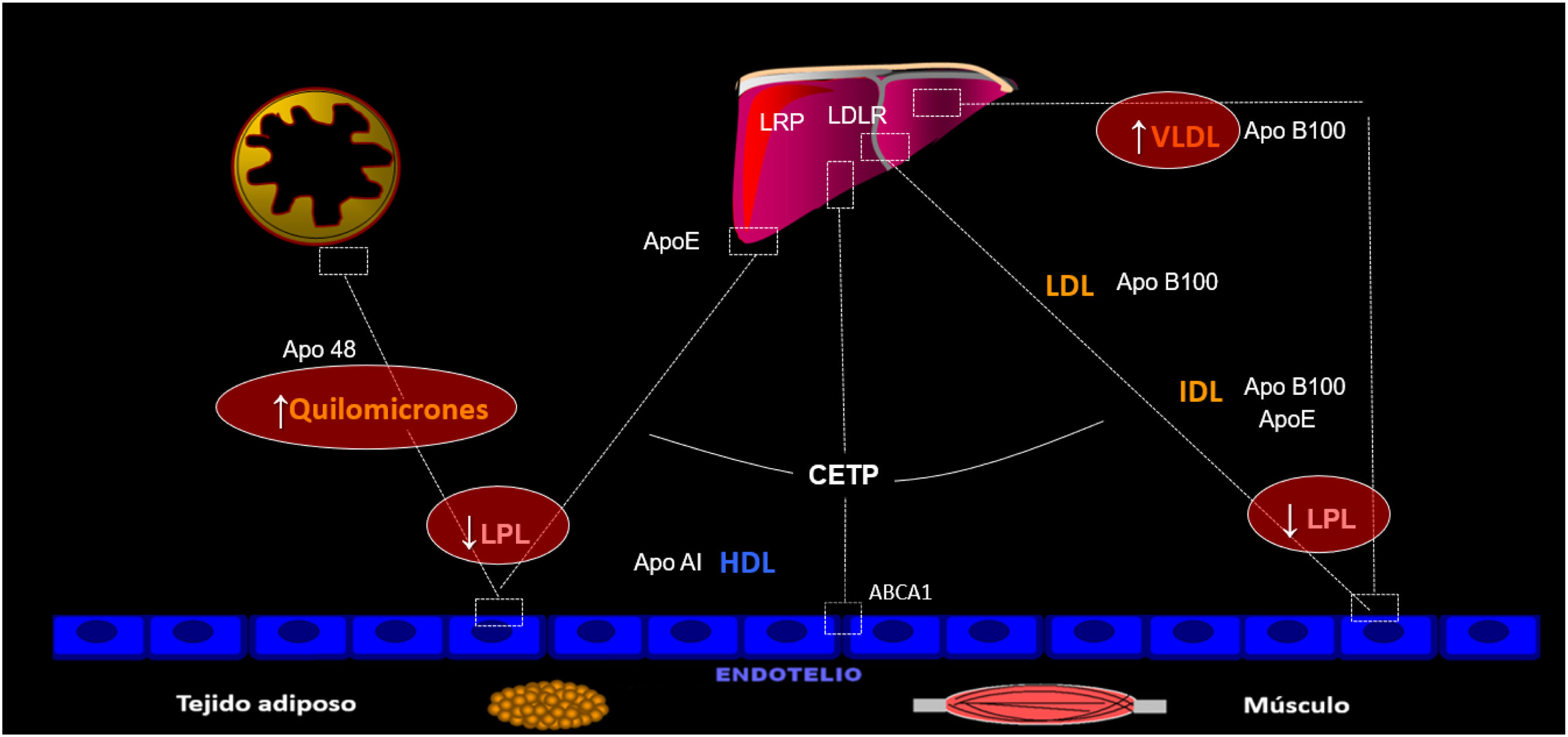
Para que esto ocurra, tiene que estar comprometido el aclaramiento plasmático de lipoproteínas ricas en triglicéridos (LRTGL), eso es, QM y lipoproteínas de muy baja densidad (VLDL), cuyo sustrato etiopatogénico común reside en un déficit funcional de lipoproteinlipasa (LPL) congénito o adquirido, parcial o total4. El aclaramiento de LRTGL es saturable, lo que ocurre con cifras de 500 a 700 mg/dL, de manera que, a partir de tales cifras de trigliceridemia, la entrada de más QM o VLDL (exceso de producción) en el torrente circulatorio no puede metabolizarse y tales partículas se acumulan en la sangre (fig. 2). En este contexto, la quilomicronemia puede coexistir con acúmulo de VLDL, dependiendo de la saturación de la tasa catalítica5.
. Cuando hay un déficit parcial o total del funcionalismo de la lipoproteinlipasa (LPL) se compromete el catabolismo de las LRTGL de manera que pueden haber quilomicronemia tras ayuno con/sin aumento de VLDL, dependiendo de la causa subyacente. Todo ello se traduce en hipertrigliceridemia grave (> 1.000 mg/dL) dado el alto contenido en triglicéridos de tales partículas. VLDL: lipoproteínas de muy baja densidad.")
Metabolismo de lipoproteínas ricas en triglicéridos (LRTGL). Cuando hay un déficit parcial o total del funcionalismo de la lipoproteinlipasa (LPL) se compromete el catabolismo de las LRTGL de manera que pueden haber quilomicronemia tras ayuno con/sin aumento de VLDL, dependiendo de la causa subyacente. Todo ello se traduce en hipertrigliceridemia grave (> 1.000 mg/dL) dado el alto contenido en triglicéridos de tales partículas.
VLDL: lipoproteínas de muy baja densidad.
La complicación clínica más frecuente, grave y característica del SQM es la pancreatitis aguda, a menudo recurrente. El riesgo de pancreatitis aumenta con hipertrigliceridemia > 1.500 a 2.000 mg/dL6. Según un reciente metaanálisis, la pancreatitis asociada a SQM tiene mayor tasa de complicaciones y morbimortalidad que la normotrigliceridémica7.
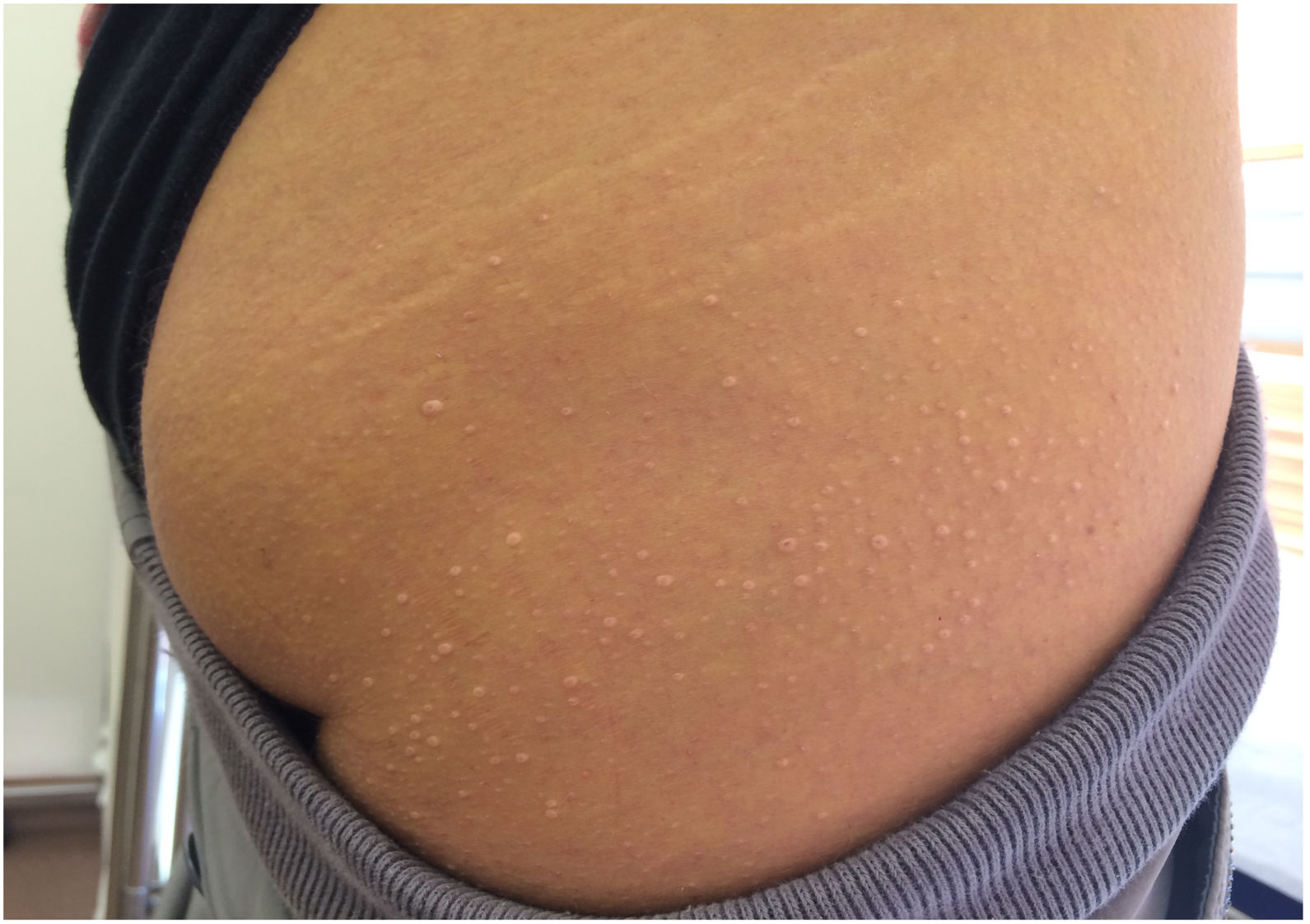
Así pues, en la valoración clínica de un paciente con cifras de triglicéridos >1.000 mg/dL8, debemos indagar sobre la historia de dolor abdominal recurrente, pancreatitis aguda y presencia de xantomas eruptivos, que pueden ser intermitentes y asociarse a períodos con cifras de triglicéridos más elevadas. Los xantomas eruptivos (fig. 3), sugestivos (no patognomónicos) de quilomicronemia familiar (QMF), suelen presentarse como pápulas amarillentas de 1 a 4 mm de diámetro rodeadas de un halo eritematoso, distribuidas más frecuentemente en glúteos y superficies extensoras de miembros (codos y rodillas)9. A veces, pueden coalescer en placas de mayor tamaño que se denominan xantomas tuberoeruptivos. En la exploración física, debemos de descartar la presencia de bocio, depósitos lipídicos extravasculares y visceromegalias. La hepatomegalia es relativamente frecuente en la esteatosis hepática que acompaña a muchas condiciones clínicas que cursan con hipertrigliceridemia, pero la esplenomegalia debe orientarnos a la presencia de QMF. Cuando sea posible, valoraremos el fondo de ojo para descartar lipemia retinalis, poco prevalente y en general relacionada con niveles sostenidos de triglicéridos superiores a 5.000 mg/dL.

En cuanto a las pruebas complementarias a solicitar en estos casos, deben incluir una analítica básica con niveles de glucosa y HbA1c, función renal, enzimas hepáticas y hormona tirotropa (TSH), así como elemental de orina con albuminuria, para el despistaje de causas secundarias de hipertrigliceridemia. El perfil lipoproteico es el primer escalón de estudio de una hipertrigliceridemia pero, cuando sea posible, la ultracentrifugación del plasma nos permitirá caracterizar con precisión el nivel de QM. Es útil reservar suero a 4 a 6°C para valorar a la mañana siguiente de la extracción el fenotipo mediante la turbidez y la presencia o no de sobrenadante cremoso (test de Havel; fig. 1). Si sospechamos quilomicronemia primaria (familiar) requeriremos el concurso de un laboratorio especializado para valorar la actividad de la LPL post-heparina y, en caso de que esté ausente o severamente disminuida, un estudio genético para secuenciación de los genes candidatos (tabla 1), aunque el orden de estas pruebas puede invertirse por razones operativas10.
Causas de quilomicronemia familiar
| • Déficit familiar de LPL |
| • Déficit familiar de GPIHBP1 |
| • Déficit familiar de Apo A5 |
| • Déficit familiar de Apo C2 |
| • Déficit familiar de LMF1 |
Apo A5: apolipoproteína A5; Apo C2: apolopoprotenína C2; GPIHBP1: glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein1; LMF1: factor 1 de maduración de lipasas; LPL: lipoproteinlipasa.
Con todo esto, los escenarios clínicos en los que puede aparecer un SQM son tres; a saber, la QMF, la quilomicronemia multifactorial (QMMF) y lipodistrofia parcial familiar (LPF), este último en los tipo 1 (síndrome de Köbberling) y 2 (enfermedad de Dunnigan) debido a un estado común de insulinorresistencia11.
Quilomicronemia familiarLa QMF es causa de SQM en individuos con quilomicronemia, pancreatitis desde edades tempranas e hipertrigliceridemia grave en ayunas (>1.000 mg/dL) como consecuencia de la existencia de mutaciones que, con carácter autosómico recesivo, afectan al gen de la propia LPL o a alguna de las proteínas que la regulan (tabla 1)12.
Así pues, la QMF incluye cinco trastornos hereditarios indistinguibles clínicamente y de manejo clínico idéntico.
Déficit familiar de LPL (MIM 238600)Es un trastorno muy poco frecuente, con una prevalencia desconocida pero probablemente en torno a 1 a 9/106, según los últimos datos conocidos. Se debe a la presencia de mutaciones (más de 220 distintas) en el gen de la LPL (8p22), siendo responsable de más del 80% de los cuadros de QMF. El patrón de herencia es autosómico recesivo, por lo que la enfermedad solo la presentan los homocigotos y, en algunos casos, los heterocigotos compuestos13-15. Los análisis de segregación familiar pueden aclarar el estatus de cada miembro de la familia y son de utilidad para el consejo genético.
La deficiencia familiar de LPL generalmente se presenta en la primera infancia (a veces al inicio de la lactancia) y se caracteriza por una hipertrigliceridemia grave con episodios de dolor abdominal, pancreatitis aguda recurrente, xantomas cutáneos eruptivos y hepatoesplenomegalia. En niños y adolescentes, se produce una elevación severa de los triglicéridos plasmáticos (1.500 a 10.000 mg/dL) en forma de QM (fenotipo I), con cifras generalmente normales de VLDL que pueden elevarse en la edad adulta (fenotipo V).
La complicación más grave y que condiciona la morbimortalidad es el desarrollo de episodios recurrentes de pancreatitis aguda. Los xantomas eruptivos suelen predominar en nalgas y extremidades y, si se busca, podemos detectar lipemia retinalis, alteración que no compromete la visión. Puede presentarse neuropatía periférica de predominio sensitivo y alteraciones neuropsiquiátricas reversibles que incluyen deterioro cognitivo y depresión. Esta entidad salvo en contadas excepciones, no parece dar lugar a aterosclerosis precoz.
Las mutaciones presentes reducen o eliminan la actividad de la LPL, evitando que la enzima hidrolice eficazmente los ácidos grasos de los QM, permaneciendo estos en el torrente circulatorio tras el ayuno, lo que se traduce en hipertrigliceridemia grave. El diagnóstico se establece con la medición de la actividad lipolítica del suero, basal y tras la administración de heparina, y las pruebas moleculares para la identificación de las mutaciones en el gen de LPL. Dado el avance de las técnicas de secuenciación genética puede invertirse el orden de las pruebas y el diagnóstico de deficiencia de LPL puede comprobarse ya con pruebas genéticas moleculares mediante la identificación de variantes bialélicas4,10.
El cuadro clínico responde a la restricción estricta de la grasa total de la dieta a < 15 a 20 g/día (d). El tratamiento nutricional, muy restrictivo y constante, mantiene la concentración de triglicéridos en plasma por debajo de 1.000 mg/d, lo que previene el dolor abdominal recurrente y reduce el riesgo de pancreatitis. La restricción de la grasa de la dieta al 15% de la ingesta total de energía suele ser suficiente para disminuir la concentración de triglicéridos en plasma y para mantener al paciente libre de síntomas, aunque es difícil de sostener en el tiempo. La grasa debe ser sustituida por triglicéridos de cadena media, los cuales se absorben directamente a la circulación portal, sin formar QM. Además, hay que ofrecer suplementos con vitaminas liposolubles (A, D, K, E). Por último, se recomienda evitar fármacos y circunstancias que aumenten la trigliceridemia, como la «huida» al consumo de hidratos de carbono, típica de las dietas muy hipograsas.
Cabe recordar que los hipolipemiantes convencionales que se usan para tratar otros trastornos del metabolismo lipídico tienen escasa o nula efectividad en personas con déficit familiar de LPL. La terapia génica fue la primera autorizada por parte de la Comisión Europea en 2012, mediante la administración de alipogen tiparvovec (Glybera®) se perseguía introducir un virus modificado para codificar el gen de la LPL, pero su elevado coste y falta de eficacia a medio-largo plazo impidieron su uso. A fecha de hoy, la única terapia farmacológica eficaz y aprobada es volanesorsen (Waylivra®)16, un oligonucleótido anti apolipoproteína CIII (Apo CIII) que ha demostrado no solo disminuir significativamente los niveles de triglicéridos sino el número de episodios de pancreatitis en la población expuesta, tanto en ensayo clínico (APPROACH)17 como en el inicio de práctica clínica real.
Es especialmente importante el seguimiento de las mujeres embarazadas con deficiencia de LPL, recomendándose una restricción de grasa extrema a < 2 g/d durante el segundo y tercer trimestre del embarazo con una estrecha vigilancia de la concentración de triglicéridos en plasma, valorándose aféresis en caso necesario.
En los episodios de pancreatitis aguda, el ayuno tiene un papel primordial debido a que favorece el rápido aclaramiento de los QM, cuya fuente principal es la ingesta de grasas. Dentro de las opciones terapéuticas, están la insulina, heparina y la plasmaféresis. La insulina y la heparina de fácil disponibilidad inducen actividad de la LPL ligada al endotelio; adicionalmente, la heparina moviliza esta enzima desde el endotelio al plasma y la insulina favorece la degradación de los QM. La plasmaféresis se ha descrito en las formas agudas, no quedando claro su papel para uso crónico dada la rápida reacumulación de QM tras las sesiones.
Déficit familiar de Apo CII (MIM 207750)Tanto el déficit de LPL como de Apo CII se presentan clínicamente en la infancia como episodios repetidos de dolor abdominal y pancreatitis aguda inducida por la quilomicronemia. Apo CII es un activador de LPL de manera que, al igual que en el déficit familiar de LPL, la actividad de LPL in vitro en plasma postheparina está disminuida. Sin embargo, en los pacientes con déficit familiar de Apo CII, la actividad lipolítica se normaliza tras la adición de plasma fresco normal que aporta la cantidad necesaria de Apo CII para que pueda realizarse la lipolisis mediada por la LPL18.
Déficit familiar de APO A5 (MIM 606638)La apolipoproteína A5 (Apo A5) es un determinante importante de las concentraciones plasmáticas de triglicéridos, debido a su papel estructural en el colesterol ligado a lipoproteínas de alta densidad (HDL) y en los QM, así como su papel en la activación de la LPL. Se expresa fundamentalmente en el hígado donde se secreta a la circulación ligada a las HDL, VLDL y a QM19.
A la luz de los conocimientos actuales, parece que Apo A5 reduce la concentración sérica de triglicéridos por estimulación de la LPL y por inhibición de la producción de VLDL en el hígado. Así, se ha sugerido que Apo A5 desempeña un papel importante en la hidrólisis de los componentes lipídicos de las lipoproteínas, siendo esta proteína clave en la unión a los proteoglicanos de la membrana plasmática de la célula endotelial, permitiendo así la actividad lipolítica endotelial20.
El gen de la Apo A5 es un candidato muy relevante en el estudio de las concentraciones plasmáticas de triglicéridos del que se han descrito más de 15 variantes potencialmente patogénicas que han mostrado asociaciones consistentes con los niveles de trigliceridemia en ayunas21.
Déficit familiar de LMF1 (MIM 246650)El factor 1 de maduración de lipasas (LMF1) es una proteína transmembrana tipo III ubicada en el retículo endoplásmico (RE), el sitio de ensamblaje y plegamiento tanto de la LPL como de la lipasa hepática (LH). Se conocen más de 20 variantes genéticas del gen de LMF1, la mayoría de significado incierto y otras que, cuando se heredan en homocigosis, determinan quilomicronemia. En humanos, la mutación más estudiada (Y439X) implica la pérdida de la fracción carboxi-terminal de uno de los dominios orientados hacia el lumen del RE que asocia una pérdida de función de LMF1 con una falta de maduración postraduccional de un alto porcentaje de LPL (superior al 90%) por plegamiento incorrecto. La LPL es retenida en el RE y finalmente degradada, lo que implica una abolición virtual de su función22,23.
Déficit familiar de GPIHBP1 (MIM 615947)La glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein1 (GPIHBP1) es una proteína clave en el proceso de cesión de ácidos grasos de los QM a los que capta y une a la LPL en el borde luminal del endotelio de la circulación capilar, funcionando como una plataforma lipolítica clave24. Se expresa fundamentalmente en el tejido adiposo, el musculo esquelético y el miocardio. Estudios recientes demuestran que además de esa función, GPIHBP1 tiene una segunda función de transporte de LPL desde su lugar de síntesis (adipocitos y células musculares), a través del intersticio y las propias células endoteliales hasta la luz capilar25. Las mutaciones deletéreas en homocigosis de GPIHBP1 constituyen la segunda causa más frecuente de QMF. Así, en el análisis genético del registro de la Sociedad Española de Arteriosclerosis (SEA), publicado en 201826, de 26 pacientes estudiados, 23 eran homocigotos de los que 19 lo eran para mutaciones de LPL y cuatro para mutaciones de GPIHBP1, incluyendo una no descrita previamente. En este registro, que incluye el mayor número de pacientes publicado en Europa hasta la fecha, no se encontraron homocigotos para Apo CII, Apo A5 ni para LMF1, aunque sigue alimentándose con nuevos casos de forma prospectiva.
Quilomicronemia multifactorialLa QMMF es la causa más frecuente del SQM. Aunque su prevalencia real se desconoce (no se ha hecho ultracentrifugación de muestras en estudios epidemiológicos) se ha estimado en 1/600 en población general27. En población adulta americana del estudio NHANES (1.999 a 2.004)28, por ejemplo, el porcentaje de sujetos con trigliceridemia > 1.000 mg/dL fue del 0,4%. En el estudio de la ciudad de Copenhague, sin embargo, tomando como criterio de quilomicronemia definitiva una concentración plasmática de triglicéridos mayor de 10 mmol/L (885 mg/dL), su prevalencia fue del 0,03% en mujeres y 0,14% en varones29.
Desde un punto de vista etiopatogénico, se requiere que, sobre una predisposición genética poligénica (una o varias variantes en más de 30 genes distintos) con bajo impacto sobre el metabolismo de las LRTGL y los niveles de triglicéridos plasmáticos, actúen factores precipitantes que incidan negativamente sobre el aclaramiento plasmático de tales lipoproteínas30. El mecanismo precipitante más frecuente sería un exceso en la producción hepática de VLDL31 que saturaría una maquinaria lipolítica ya de por sí comprometida4. El resultado es la presencia de QM y elevada cantidad de VLDL en una muestra de sangre obtenida tras ayuno y cuyo suero refrigerado (4°C durante 16 a 18 h) se correspondería con el fenotipo V de la clasificación de Fredrickson2 (fig. 1). No todos los pacientes, por tanto, con predisposición genética para SQM lo desarrollarán. El estudio de las familias ha permitido observar que los casos índices, a menudo diagnosticados previamente de hipertrigliceridemia familiar o hiperlipemia familiar combinada, tienen niveles mucho más elevados de trigliceridemia que sus familiares de primer grado con hipertrigliceridemia32.
Las causas precipitantes o secundarias de QMF33 se recogen en la tabla 2, siendo lo más frecuente la coexistencia de diabetes y obesidad29,34.
Causas precipitantes de quilomicronemia multifactorial
| Condiciones clínicas | Diabetes (1 y 2) no conocida o mal controlada |
| Obesidad | |
| Hipotiroidismo | |
| Enfermedad renal crónica | |
| Síndrome nefrótico | |
| Embarazo | |
| Lupus eritematoso sistémico | |
| Drogas y fármacos | Alcohol |
| Cardiovasculares: β-bloqueantes, diuréticos (tiacídicos y de asa) | |
| Hormonas: glucocorticoides, estrógenos, tamoxifeno y raloxifeno | |
| Inmunosupresores: ciclosporina, tacrolimus, sarolimus | |
| Psicofármacos: mirtazapina, olanzapina | |
| Secuestradores de ácidos biliares | |
| Inhibidores de la proteasa del VIH | |
| Agonistas de los receptores X retinoides: ácido retinoico | |
| Propofol |
Un aspecto de interés práctico ante un SQM, es el diagnóstico diferencial entre QMF y QMMF. Desgraciadamente, no hay ningún dato clínico que nos permita diferenciar ambos procesos de manera inequívoca. Con esa premisa, la tabla 3 recoge una serie de datos que pueden ayudar para este fin35,36 con el propósito de optimizar las recomendaciones sobre hábitos de vida y la selección de candidatos para el estudio molecular definitivo.
Aspectos clínicos diferenciales entre QMF y QMMF
| QMF | QMMF | |
|---|---|---|
| Lipoproteína predominante | QM (fenotipo I) o QM + VLDL (fenotipo V) | QM + VLDL (fenotipo V) |
| Aspecto del suero | Lipémico | Lipémico |
| Aspecto suero tras refrigerado | Sobrenadante lechoso e infranadante traslúcido o sobrenadante lechoso e infranadante turbio | Sobrenadante lechoso e infranadante turbio |
| Historia de pancreatitis | ++++ | ++ |
| Obesidad | ± | ++++ |
| Historia de diabetes | + | ++++ |
| Hiperlipemia familiar combinada | + | ++++ |
| Historia de trigliceridemia < 500 mg/dL | ± | +++ |
| Respuesta a fibratos | - | + |
QM: quilomicrones; QMF: quilomicronemia familiar; QMMF: quilomicronemia multifactorial; VLDL: lipoproteínas de muy baja densidad.
En esta dirección, un grupo de expertos37 ha propuesto un «score pragmático» para la identificación de pacientes con QMF, basándose en ocho ítems clínico-biológicos (inicio en la juventud, hipertrigliceridemia persistente > 880 mg/dL, historia de pancreatitis/dolor abdominal, exclusión de causas secundarias o de hiperlipemia familiar combinada y ausencia de respuesta a fibratos). Una puntuación ≥ 10 puntos en dicho score, hace muy probable el diagnóstico de QMMF.
La identificación y corrección de los factores precipitantes de SQM constituyen el pilar del tratamiento en la QMF. Además, en estos casos, la terapia concomitante con fibratos puede ayudar a corregir la hipertrigliceridemia.
Conflicto de interesesLos autores declaran no tener conflictos de interés con el contenido del presente trabajo.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.