



Muscle residual force enhancement has been observed in different muscle preparations for more than half a century. Nonetheless, its mechanism remains unclear; to date, there are three generally accepted hypotheses: 1) sarcomere length non-uniformity, 2) engagement of passive elements, and 3) an increased number of cross-bridges. The first hypothesis uses sarcomere non-homogeneity and instability to explain how “weak” sarcomeres would convey the higher tension generated by an enhanced overlap from “stronger” sarcomeres, allowing the whole system to produce higher forces than predicted by the force-length relationship; non-uniformity provides theoretical support for a large amount of the experimental data. The second hypothesis suggests that passive elements within the sarcomeres (i.e., titin) could gain strain upon calcium activation followed by stretch. Finally, the third hypothesis suggests that muscle stretch after activation would alter cross-bridge kinetics to increase the number of attached cross-bridges. Presently, we cannot completely rule out any of the three hypotheses. Different experimental results suggest that the mechanisms on which these three hypotheses are based could all coexist.
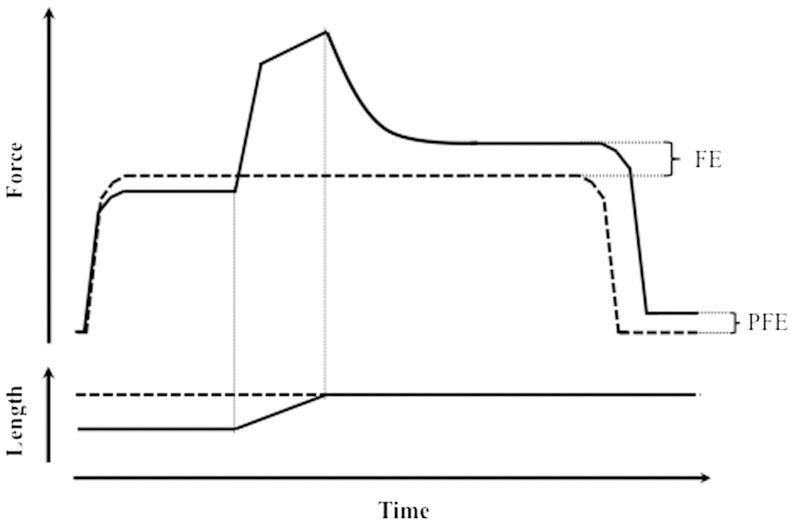
When a muscle is stretched while activated and held at a certain final length long enough for the force transients to cease, the steady force achieved is always higher than the steady force that develops when the muscle is activated while already held isometrically at the same final length (1-5) (Figure 1). This residual force enhancement was first observed by Abbott & Aubert (1) and predated Huxley's cross-bridge model (6). Nevertheless, this enhancement could not be accounted for by either this model (6,7) or the force-length relationship (8); to date, the mechanism of force enhancement remains elusive.
, and then isometrically activated and kept at the same final length before being relaxed again (dashed lines). Top panel: force traces. Bottom panel: length traces. FE: force enhancement. PFE: passive force enhancement.")
Schematic representation of muscle residual force enhancement. Both panels show two representative superimposed contractions from the same muscle as it is first activated, stretched to a certain final length, relaxed (solid lines), and then isometrically activated and kept at the same final length before being relaxed again (dashed lines). Top panel: force traces. Bottom panel: length traces. FE: force enhancement. PFE: passive force enhancement.
Residual force enhancement has been observed in human muscle during electrical and voluntary stimulation (9,10), in isolated muscle fibers (11-14), in myofibrils (4,15,16) and even in single sarcomeres (17). Force enhancement is known to be long lasting and to increase with increasing stretch magnitudes (1,2,11,14,18), but it is also independent – or almost independent (19) – of stretch velocity (1,19). Additionally, it is decreased but not fully abolished after deactivation (10,18).
Several possible explanations for residual force enhancement have been proposed, although none are able to fully elucidate its mechanism (3). This paper provides a brief review of the three generally accepted hypotheses for residual force enhancement: 1) sarcomere length non-uniformity, 2) engagement of a passive element, and 3) increase in the number of cross-bridges. Our prime goal is to recapitulate and summarize these three hypotheses for residual force enhancement in a more digestible manner, allowing a wider audience to understand a phenomenon that can also be relevant to other fields, such as exercise physiology, biomechanics, healthy-gait mechanics, physical therapy, etc. If more in-depth information is needed, we suggest the following reviews to cover specific aspects of residual force enhancement: Herzog et al. (3), Rassier and Herzog (20), Edman (21), and Rassier (22).
Hypothesis 1 - Sarcomere length non-uniformitySarcomere length non-uniformity was the first mechanism used to explain force enhancement after stretch (23), being still currently accepted and advocated (21). The first study to present a possible explanation and indirect evidence that sarcomere length non-uniformity was involved in residual force enhancement was performed by Julian and Morgan (24). Later, Morgan (25) provided a more complete explanation that could account for most of the residual force enhancement characteristics found experimentally. One of the bases for non-uniformity within sarcomeres is the phenomenon of instability on the descending limb of the force-length relationship (26). An unstable system allows differences in individual sarcomere lengths: some sarcomeres elongate more, becoming weaker (due to decreasing filament overlap), and others in turn become even stronger (due to increasing overlap). The “weaker” sarcomeres continue to elongate until a critical point at which they “yield;” in other words, the “weaker” sarcomeres' cross-bridges can no longer maintain adequate tension, which leads to their detachment (yield). These sarcomeres continue to elongate until the tension borne by passive (non-contractile) elements equals the tension of the “strong” sarcomeres, and in this case, the “weak” sarcomeres are “popped.” The remaining tensile force is now determined by the stronger sarcomeres, which “transmit” their tension through the “popped” sarcomeres, thereby enabling the entire system to exhibit greater force than would be predicted by the average sarcomere force based on the force-length relationship curve (25).
Support for the theorySeveral studies (1,24,27) have shown that force enhancement occurs only on the descending limb of the force-length curve, which is believed to be unstable (26). In support of this idea, Morgan (25) showed that in the absence of instability, sarcomeres would theoretically stretch uniformly, and no enhancement should be expected.
Sarcomere length non-uniformities may also provide an explanation for different force-enhancement behaviors under differing conditions, such as velocity independence of force enhancement and stretch-magnitude dependence (1,2). Muscle-stretch velocity neither prevents nor affects instability (25,26); therefore, sarcomere length non-uniformity must be independent from stretch velocity. In contrast, force enhancement should be directly related to stretch magnitude because the more a muscle is stretched, the greater the number and degree of sarcomere elongation and greater the production of residual tension. A mathematical explanation has been provided for this behavior (25).
What Julian and Morgan (24) noted in their study was that sarcomeres from the centers of the cells stretched more than sarcomeres near the ends. Talbot and Morgan (28) confirmed this observation. Using electron microscopy, they were able to observe that certain half-sarcomeres were more elongated than the average half-sarcomere.
One of the strongest pieces of evidence presented by sarcomere-non-uniformity advocates is the decrease in stiffness. Assuming that the number of formed cross-bridges is the main factor responsible for increased muscle stiffness, both longer and “popped” sarcomeres should each present a lower stiffness than the shorter sarcomeres. Therefore, stiffness should decrease after a muscle is stretched. This expected decrease in stiffness has been reported in some studies (29-31).
Limitations and criticismsThe idea of instability has long been one of the bases for explaining sarcomere length non-uniformity. This notion was explored by Morgan (25) from Hill's (26) idea of dynamic instability derived from the negative slope on the descending limb of the force-length curve. Conceptually, this approach was inadequate because one cannot assume dynamic instability from a construct based on observations made under isometric conditions (3). However, even when the sarcomeres were clamped and kept stable to prevent non-uniformities, force enhancement was still observed (1). More recently, results obtained using myofibril preparations showed that although sarcomeres did not stretch uniformly on either the descending (32) or ascending (4) limb of the force-length curve, they remained stable after stretch, indicating that force enhancement can occur even in the absence of instability.
A number of studies have shown force enhancement on the supposedly stable ascending limb of the force-length curve. These studies were performed not only in fibers (13,33) but also in muscles in situ (9,10,18) and even in myofibrils (4). All of these studies reported small but consistent force enhancements on the ascending limb.
According to the sarcomere length non-uniformity theory (25), force enhancement after stretch should not be higher than the tension produced at the initial length, let alone exceed the force at the optimal sarcomere length (plateau). Again, several experiments performed with different muscle preparations have refuted this assumption and shown that force enhancement could exceed the maximal isometric force developed at the plateau of the force-length relationship (4,10,13,15),.
In contrast to studies that have found decreased stiffness in the force-enhanced state (29-31), other studies have found an increase in stiffness when comparing the isometric reference force with the force in the enhanced state in different muscle preparations (34,35). Clearly, controversy remains regarding the explanation for stiffness behavior after stretch.
Finally, the development of techniques that have enabled the investigation of isolated myofibrils (36) and even single sarcomeres (37) has brought further skepticism regarding the sarcomere length non-uniformity theory. Studies have found force enhancement not only while sarcomere lengths were kept stable within activated myofibrils that were stretched (4,15) but also in isolated single sarcomeres (17). Both studies rule out sarcomere length non-uniformity as an explanation for force enhancement. For non-uniformity to be accepted, this concept must be extended to include half-sarcomere length non-uniformity (22). A-band displacements in preparations with myofibrils (38,39) and individual sarcomeres (37) were clearly detected during and after activation. There has been increasing evidence that residual force enhancement could be attributed to asymmetrical length changes inside one sarcomere that caused an increasing overlap in one of the two halves (21). A recent mathematical model (40) using a series of dissimilar half-sarcomeres (in several conditions) was able to generate a residual force enhancement on the order of 13% above the isometric reference contraction. Although exciting, this simulation was not able to produce higher levels of force enhancement or a duration longer than 30 s. According to the model, when both halves cease to lengthen or shorten, the different populations of cross-bridges reach an equilibrium, causing the force to decrease to the same level as the isometric reference. Hence, half-sarcomere length non-uniformity cannot fully account for the residual force enhancement. It is possible that simulations with more myofibrils in parallel could lead to a further increase in the magnitude and duration of the force enhancement found.
Hypothesis 2 – Engagement of a passive elementIt has been suggested that force enhancement after stretch may also be related to an increase in strain in passive elastic elements inside the muscle (12). These elements may not, in fact, be totally “passive;” they become stiffer when muscles are stretched while activated. The most likely “passive element” for this function is titin (18).
Support for the theoryA series of studies (14,18,19,41) demonstrated an increase in the passive force even after the muscle was relaxed from the enhanced state (Figure 1). This was called passive force enhancement. Generally speaking, this behavior is very similar to the actual total force enhancement; it is velocity independent, increases with stretch magnitude and is persistent (18,41,42). However, its magnitude is smaller than the total enhancement (18,42) and is thus assumed to account for only part of the phenomenon.
Previously, the origin of passive force enhancement was unknown, but it was commonly interpreted as the engagement of a passive element that becomes stiffer after stretch during activation and is not eliminated by deactivation (3,18). Passive and total force enhancements were shown to diminish when a shortening is applied prior to stretch, which was attributed to a release in the strain on elastic structures whose force was enhanced by the stretch (14). As mentioned previously, titin is considered the most likely candidate for this stretch-activation-sensitive role (16-18,43), because its stiffness can increase due to the effect of calcium binding to two specific motifs, the PEVK (P-proline, E-glutamine, V-valine, K-lysine), and immunoglobulin (Ig) domains, that have been found to change their molecular conformation upon calcium activation (45) and through binding to actin (46).
Increasingly, researchers have suggested that passive force enhancement may actually play a greater role than previously thought in contributing to the total residual force enhancement (18,42). It is believed that the greater the stretch magnitude, the greater the contribution of passive forces to the total force enhancement (18,42). Herzog and Leonard (18) found a greater than 80% passive force enhancement contribution to the total force enhancement at longer lengths. Even on the ascending limb of the force-length relationship, where passive forces are not thought to play a significant role, a small passive force enhancement was detected (47). Indeed, Leonard and Herzog (44) recently showed that passive forces could be increased to almost 400% when relaxed myofibrils were compared with activated myofibrils while having their average sarcomere-length stretched from 2.4 μm to 6.0 μm. The authors attributed this notable increase in force to a combination of two mechanisms: 1) titin stiffening mediated by calcium activation and 2) a progressive binding of titin to actin through a mechanism that is likely to be force dependent and elicited by stretch after activation. This interaction between titin and actin was not a novel observation, but Leonard and Herzog (44) proposed a new understanding for its role in residual force enhancement. The authors also suggested that the high levels of force enhancement encountered could confer a protective mechanism against stretch-induced muscle injuries.
Limitations and criticismsPassive forces can be modulated upon calcium activation (48), showing that an increase in stiffness can be cross-bridge independent (49). As previously mentioned, this increase in stiffness is attributable to titin (18). Based on these results, it is assume that at least the passive force enhancement could be attributed to titin activation. However, titin stiffening via calcium binding only accounts for 25% of the total passive force enhancement (43). Therefore, calcium-induced titin stiffening alone is insufficient to explain even the passive part of force enhancement.
The explanation proposed by Leonard and Herzog (44) is attractive but has been criticized (50); although one study showed that titin can interact with actin (51), it remains unclear whether there is in fact interaction between PEVK and Ig and actin. It may be that stretches beyond the sarcomere length of 4.0 μm could have the effect of unfolding these domains and allowing their interaction with actin, but there is little evidence for this possibility (22,50).
The levels of passive force observed by Leonard and Herzog (44) do not agree with results from a similar study that also reported passive force modulated by a length-dependent combination of stretch and activation (48). In this study, the greatest passive force increases found were approximately 30%. However, care should be taken when comparing results from these two studies because the latter used single fibers instead of myofibrils, and their average sarcomere length did not exceed 3.2 μm. Still, a 400% force increase is very unlikely since myofibrils are much more fragile than fibers, which also generate significantly lower levels of passive force (36,52). Furthermore, it is difficult to grasp why muscles would have such a strong protective mechanism, as Leonard and Herzog (44) suggest, if they are rarely stretched to that extent during muscle contractions in vivo.
Hypothesis 3 – Increase in the number of cross-bridgesAnother hypothesis to explain increasing tension in the enhanced state is an increase in the number of attached cross-bridges. There are several possible mechanisms at work: a stretch-induced phosphorylation of myosin light chains (52), a disordering of the myofilament lattice (53), or an increase in the duration of attachment (dwell time) (3).
Support for the theorySeveral studies have found an increase in stiffness in the force-enhanced state compared with the isometric reference state in different muscle preparations (34,35,54). Assuming that most of the increase in muscle stiffness is due to an increase in the number of cross-bridges (55,56), these studies provide indirect evidence for our third hypothesis. For this increased stiffness to occur, according to Huxley's model (6), either the cross-bridge rate of detachment g(x) should decrease, or the rate of attachment f(x) should increase.
Stretch after activation may induce cross-bridge phosphorylation (57), myofilament-lattice disordering (53), and/or repartitioning of the cross-bridge population between the “force-bearing” and “non-force-bearing” molecular states (58-61). In the first case, the phosphorylated myosin light chains would bend myosin heads towards the actin filament (62,63). In the second case, myofilament disordering would cause the thin filaments to move closer to the thick filaments (53). In the third case, changes in cross-bridge distribution between the two aforementioned molecular states could induce an increase in the “non-force-bearing” state that would indirectly slow down the detachment rate after stretch because there would be more bridges in the “non-force-bearing” state available to move forward to the “force-bearing” state (59,60). Finally, stretch per se could increase the time of attachment by slowing phosphate and ADP release, which has been previously observed in studies of single-molecule (myosin and actin) interactions (8,64,65).
Limitations and criticismsThe studies that compare stiffness in the force-enhanced state to the corresponding isometric state are few and contradictory. There are nearly as many studies that have shown either no change (24) or a decrease (29-31) in stiffness as studies that have shown increased stiffness (34,35,54). In addition, there are limitations in using stiffness measurements to make assumptions about the number of cross-bridges, especially after stretch. Considering that this measurement would reflect the whole system's stiffness (i.e., titin, filaments, and the number of detached cross-bridges in the “non-force-bearing molecular state”), an increase in stiffness could also be attributed to an increase in strain on titin due to “popped” sarcomeres that would convey an enhanced stiffness from shorter, strong sarcomeres.
Even accepting that the number of cross-bridges is increased in the force-enhanced state due to a decrease in the myofilament space induced by stretch, which would in turn facilitate light-chain phosphorylation and force potentiation, as suggested by Edman (52), it is unlikely that stretch-induced myosin phosphorylation would be as long lasting as has been previously reported (1,3,4), which would be necessary for it to have a duration comparable to residual force enhancement. In fact, Edman (52) was concerned with characterizing force enhancement during force development; his method examined the force increase occurring right before the breakpoint on the force myogram, not after the force transients were absent. Therefore, the mechanism suggested by this author is plausible to explain these force transients, but it is unlikely to also explain residual behavior. Similarly, there is no reason to believe that stretch-induced myofilament disordering (53) or the redistribution of the cross-bridges between “non-force-” and “force-bearing” states (59,60) when considering that only the stretch effect could continue for several seconds after stretch, which is as long as the residual force enhancement has been reported (1,14,18,19). In other words, both mechanisms should be continually maintained for the cross-bridge-kinetics to remain permanently altered after the stretch, which is unlikely.
Finally, the results from studies of single myosin molecules with few myosin interactions that were observed for a brief time interval (64-66) are difficult to extrapolate to long-lasting phenomena such as residual force enhancement. This limitation is particularly true because residual force enhancement was studied in preparations at the single sarcomere level and up that involved several cross-bridge cycles occurring for many seconds after the stretch (1,4,10,13,15),.
Summary and ConclusionsControversy remains concerning the three hypotheses discussed. Sarcomere length non-uniformity has been highly criticized not only because some of its predictions have been shown to be incorrect but particularly in light of recent results from muscle preparation studies that show force enhancement even at the single sarcomere level. For non-uniformity to be considered the exclusive mechanism of residual force enhancement, the concept of half-sarcomere length non-uniformities causing force increase must be more thoroughly tested experimentally.
There is sufficient evidence showing that a passive elastic element (i.e., titin) could also be responsible for force enhancement at longer lengths. However, it is unlikely that this is the exclusive mechanism since the passive force enhancement is always smaller than the total force. In fact, the cause of passive force enhancement is still not entirely clear; experiments that indirectly evaluated titin activation mediated by stretch after activation were able to attribute only 25% of the passive force enhancement to titin activation. Furthermore, findings on passive force regulation are contradictory. Similar studies have obtained very different values for increased force magnitude.
There is some evidence showing an increase in the number of cross-bridges when comparing the force-enhanced with the corresponding isometric state. It appears that a stretch after activation can produce long-lasting changes in cross-bridge kinetics. However, the exact mechanism for this remains unclear.
Thus far, none of the three hypotheses provide a full explanation, although none can be completely ruled out. Evidence has shown that sarcomere length non-uniformity alone can no longer explain residual force enhancement. To do so, the concept must be extended to include half-sarcomere length non-uniformity as a source of force enhancement, although it is also unlikely half-sarcomere length non-uniformity alone could explain force enhancement. Therefore, based on several strong but partial pieces of evidence and the contradictions from the three hypotheses presented here, we suggest that residual force enhancement is perhaps caused by all three mechanisms operating together: i) engagement of a passive element, likely via titin activation and interaction with actin, ii) dissimilarities in half-sarcomere lengths, and iii) an increase in the number of cross-bridges due to alterations of their kinetics caused by stretch after activation.
Fábio C. Minozzo received a fellowship from Fonds de Recherche du Québec – Nature et technologies (FRQNT).
No potential conflict of interest was reported.
Minozzo FC and Lira CA contributed to manuscript preparation, interpretation, and critical revision of the manuscript.