



Pancreatic endocrine tumors (PET) are extremely rare, accounting for less than 1% of all pancreatic neoplasms and may present with a variety of clinical syndromes depending on the presence and type of hormone production.1
Pancreatic endocrine tumors include insulinomas, gastrinomas, VIPomas (tumors that secrete vasoactive intestinal peptide), glucagonomas, somatostatinomas, and nonfunctioning pancreatic endocrine tumors. Insulinoma is the most common PET, and gastrinoma is the second most common; consequently, in most cases, PET produces hypoglycemic or hypergastrinemic syndromes.1 The remaining PETs are very rare indeed.1 Thus, the presentation of these tumors with a clinical feature of diarrheogenic syndrome is an exceedingly rare clinical entity. The clinical features of VIPoma are a severe but intermittent diarrhea, often of a watery nature. A true carcinoid tumor of the pancreas accounts for less than 1% of gastrointestinal carcinoids2–9 and generally has a poor prognosis.8–11 The most frequent symptoms found in patients with pancreatic carcinoids are abdominal pain and diarrhea.5,8–11
Although the term carcinoid is well established in medical terminology, it is no longer adequate to cover the entire morphological and biological spectrum of neoplasms of the disseminated neuroendocrine cell system. Therefore, the WHO classification published in 2000 uses instead the general terms, neuroendocrine tumor and neuroendocrine carcinoma. Although the pancreas is an uncommon location for a carcinoid tumor, the complex cell population of the pancreas may give rise to a large group of endocrine tumors with pluripotential secretory capabilities.2 Pancreatic carcinoids originate from the enterochromaffin cells (Kultschitsky cells) that are usually present in the exocrine glands of the pancreatic tissue and in the pancreatic islet cells, which retain the capacity to secrete serotonergic derivatives, 6,7,12 carcinoids also originate from multipotent stem cells of the duct epithelium that may differentiate into any one of a variety of adult endocrine-secreting cells, mainly into monoamine and/or various peptide hormone-producing cells.4,13 It is apparent that the endocrine component cells of the pancreas consist not only of the cells of the islets of Langerhans, but also of endocrine cells in its ducts and acini.14
When compared with carcinoids in other sites, the pancreatic carcinoid has specific features: (i) the lowest incidence; (ii) argyrophil stain is more likely to be positive than argentaffin; (iii) tendency to be the largest; and (iiii) highest incidence of metastases and carcinoid syndrome, resulting in the most unfavorable prognosis.8
The objective of this report was to describe an additional case of carcinoid tumor of the pancreas with a clinical feature of diarrheogenic syndrome, adding to those previously reported, and to review the world literature in an attempt to define the clinical presentation, morphologic findings, treatment, and prognosis of this unusual tumor.
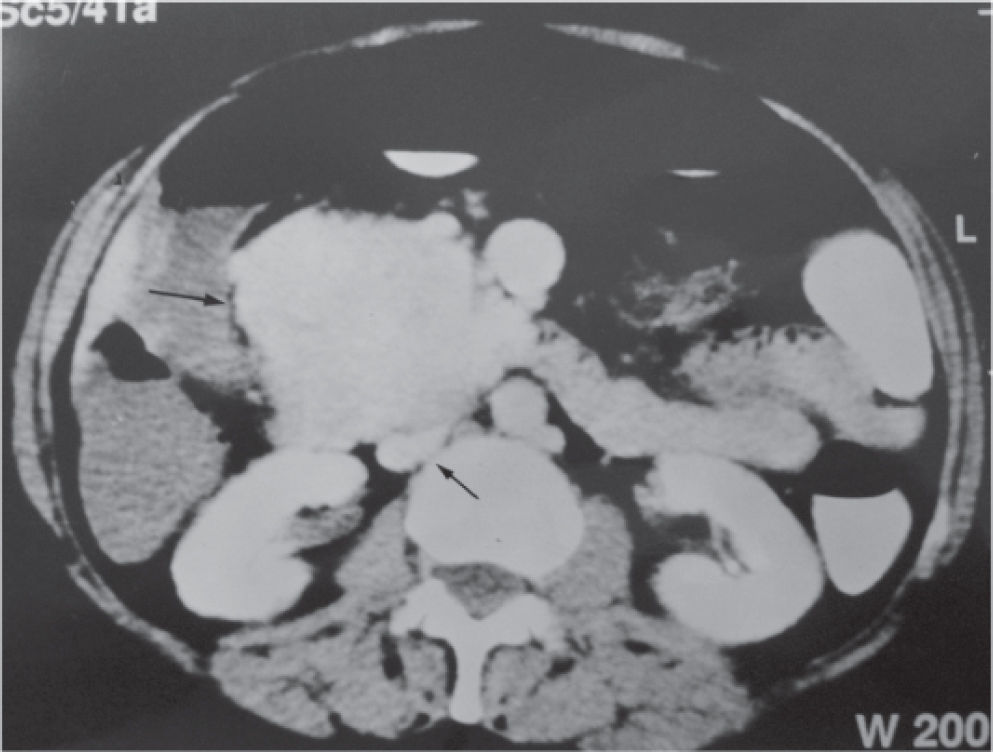
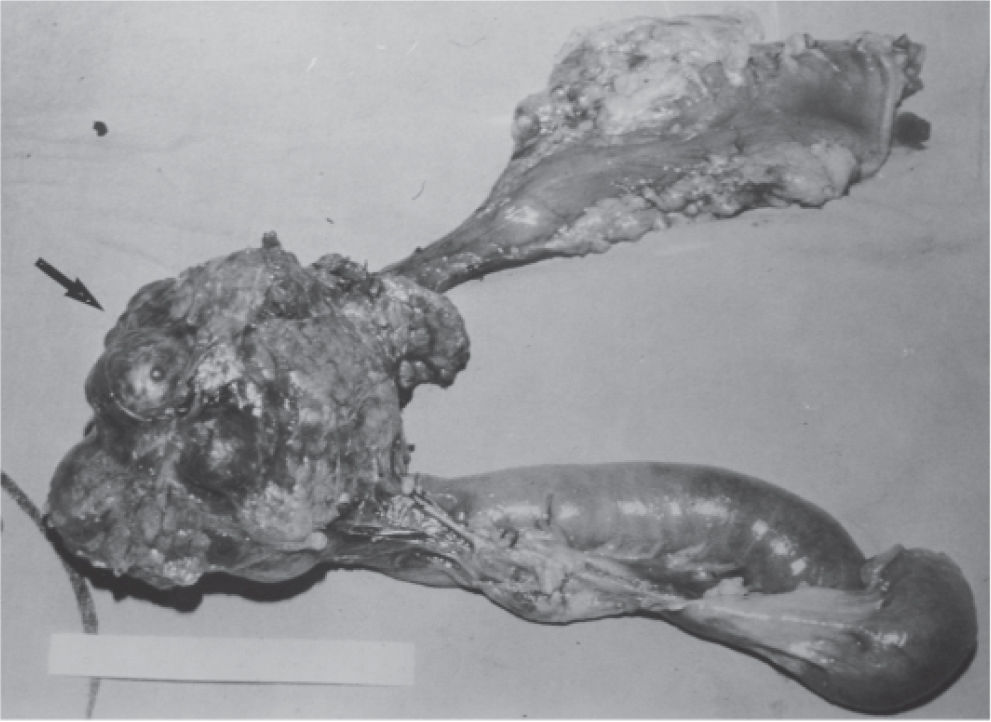
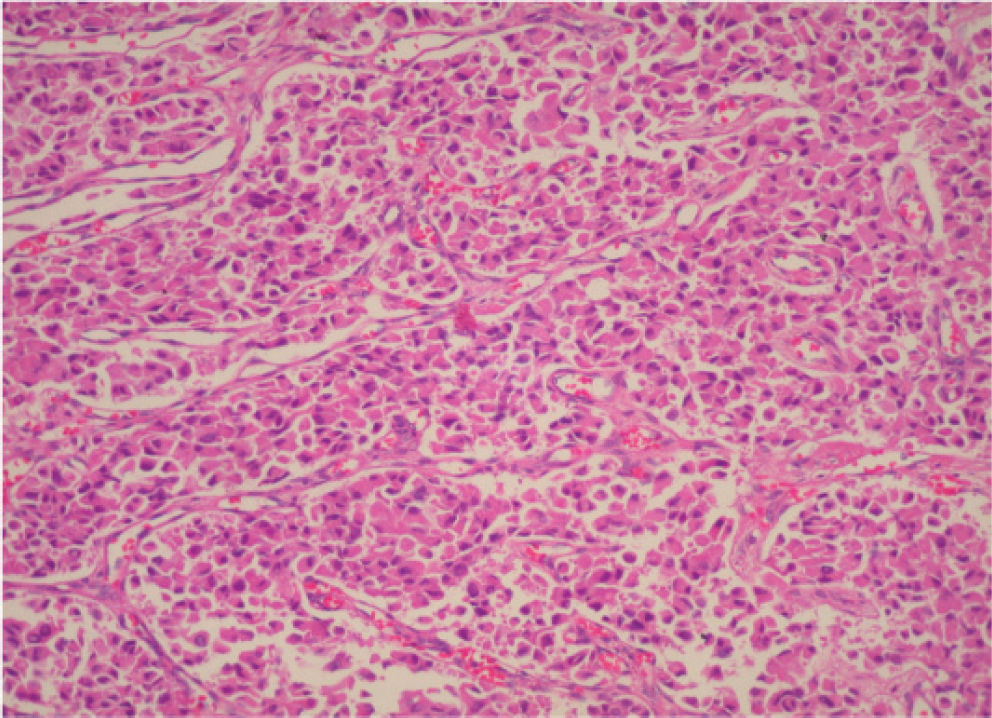
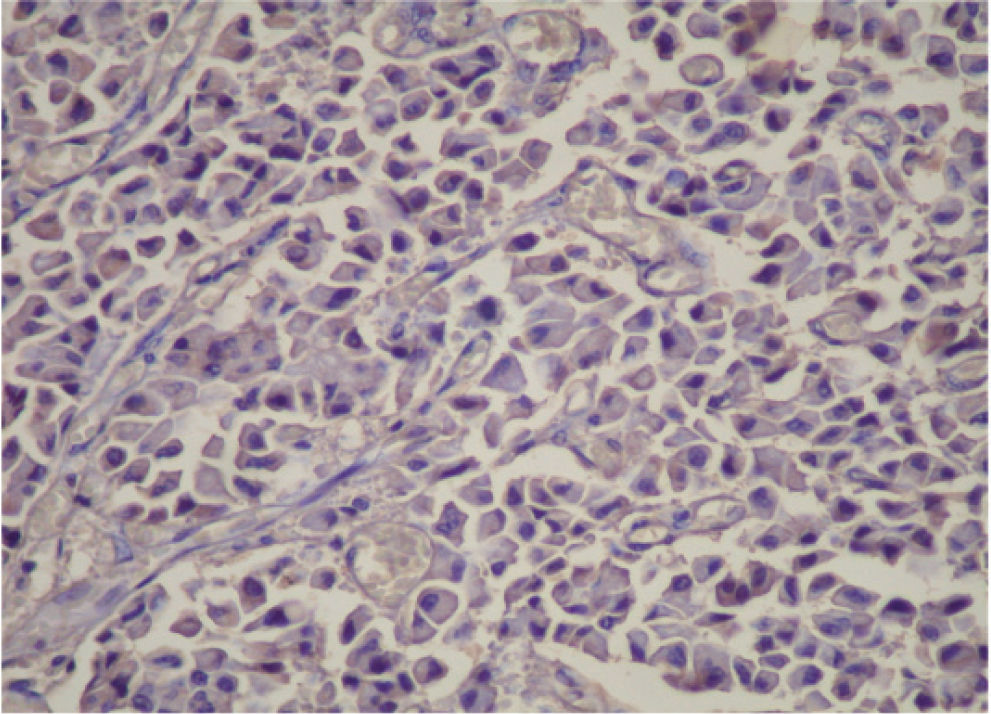
CASE REPORTA 62-year-old white woman was admitted to the Servidor Público State Hospital because of 3 to 6 evacuations per day of pasty or liquid feces with onset 2 years previously and without the presence of blood. She denied having abdominal pain or weight loss. Physical exam did not show significant alterations. The 24-hour fecal fat excretion, amylasemia, potassium, fasting glycemia, and serum values of carcinoembryonic antigen (CEA) and CA 19-9 were normal. Abdominal sonography revealed the presence of a retroperitoneal solid mass. Computerized tomography (CT) and magnetic resonance of the abdomen showed a solid and irregular mass located in the cephalic area of the pancreas, proximal to the inferior vena cava (Fig. 1). Phlebography of the inferior vena cava, performed via the femoral vein, did not reveal signs of invasion. Exploratory laparotomy detected a hardened mass in the cephalic portion of the pancreas, with an irregular surface, measuring 12 x 8 cm in its largest diameter, that was squeezed and stretched to the 1st and 2nd portions of the duodenum (Fig. 2). The liver exhibited a normal and consistent aspect, and the remaining areas of the abdominal cavity did not present alterations. The pancreatic lesion was extirpated by partial pancreatoduodenectomy (Whipple's operation). Anatomopathological exam of the surgical specimen revealed a pancreatic carcinoid tumor with angiolymphatic invasion that infiltrated the Vater's ampulla and the duodenal wall (Fig. 3). The lymph nodes, in a total of 15 structures, and the margins of the lesion were free from neoplastic involvement. Immunohistochemical studies were performed on paraffin sections using commercially available primary antibodies directed against a set of neuroendocrine antigens as well as different polypeptides. The neoplasia exhibited reactivity to neuron-specific enolase (NSE), EMA, CEA, factor VIII, serotonin (Fig. 4), glucagon, and somatostatin, and was not reactive to PS100, vimentin, cytokeratins AE1/AE3, or insulin. In the postoperative period, the patient developed venous thrombosis of the lower limbs, without other complications, and was discharged on postoperative day 15. She remains well, without symptoms, 5 years after the operation.
 in the area at the level of the head of the pancreas, showing the proximity between the mass and the inferior vena cava (small arrow)")

")
Micrograph of a histological section of the pancreatic tumor showing monomorphic tumor cells growing in an organoid pattern characterized by nests, sheets, and ribbons. The tumor cells are small, round to oval, with nuclei showing “salt and pepper” chromatin and with prominent granular eosinophilic cytoplasm. The histological pattern is consistent with a carcinoid tumor (hematoxilin & eosin staining; original magnification, X200)
")
Although the histogenesis of carcinoid tumors of the pancreas has yet to be fully clarified, embryologic studies have demonstrated that argentaffin cells are present throughout the foregut and migrate to the bronchioles, pancreatic ducts, and Langerhans islets.7,8 Wilson et al14 demonstrated immmunoreactivity to enterochromaffin cells in islet cell tumors, and in non-neoplastic ducts and ductule, acini and Langerhans islets. It has been suggested that the islet cell and argentaffin cell are derived from the same neuroectodermal precursor or from the same multipotential precursor cells of the ductal epithelium, with the capacity to proliferate and differentiate themselves from peptide or monoamine hormone-producing cells.7,8
Carcinoid tumors are immunohistochemically positive for the pan-endocrine markers, neuron-specific enolase and chromogranin A, and also for antiserotonin antibodies, which are considered specific markers for such tumors.3,4,8,9 When immunohistochemical techniques are positive for serotonin in the tumor cells, the tumor can be classified as a carcinoid tumor.3,7 In the present case, the immunohistochemical study of the neoplastic tissue exhibited reactivity to serotonin, but also to glucagon and somatostatin, among others. This fact suggests that the tumor probably originated from pancreatic islet cells. As summarized by Mao et al,8 several carcinoids of the pancreas have stained positively for other islet cell hormones.
Pancreatic neuroendocrine tumors are extremely rare in children, but occur in all age groups and in males and females with equal frequency,1 and they can occur in all parts of the pancreas.1 Pancreatic carcinoids, because of their location, tend to produce relatively vague, nonspecific symptoms resulting in larger, more advanced tumors at presentation.5,8,9
Less than 10% of carcinoids present diarrhea caused by systemic serotonin release. The explanation lies in the efficient hepatic metabolism of vasoactive amines, and that is also the reason why carcinoid syndrome rarely occurs in the absence of liver metastasis. Exceptions are circumstances in which venous blood from a large tumor was drained directly into systemic circulation11 as may have occurred in the present case.
The diarrhea associated with pancreatic carcinoids is generally a consequence of pancreatic exocrine insufficiency and or intestinal action of serotonin produced by the neoplasia.3,8,15 One might speculate that perhaps different mediators produce diarrheas of different intensities.15 Studies of the possible role of vasoactive intestinal polypeptide (VIP), prostaglandin E, and serotonin in the cause of the watery diarrhea syndrome have attracted more attention. Machado et al16 described a patient with a Verner-Morrison diarrheogenic syndrome with a high plasma concentration of VIP due to a neuroendocrine tumor located in the body and tail of the pancreas and multiple liver metastases. The diarrheogenic syndrome was controlled by surgical resection of the pancreatic neoplasm and postoperative hepatic artery embolization.
In our patient, the diarrhea was intermittent and of lesser severity than that found in VIPomas. Unfortunately, serum values for the potential mediators of humoral diarrhea are not available. Nonetheless, the results from the immunohistochemical studies were positive for serotonin, suggesting that it might be involved in causing the diarrhea syndrome.
Jaundice is an unusual finding in pancreatic carcinoids, as explained by the tumor location in the pancreatic body and tail in 70% of patients and the slow and late infiltrative growth.5,8,9 Although the lesion of the present case was situated at the head of the pancreas and presented large dimensions, there was no clinical or laboratory jaundice, probably due to the noninfiltrative nature of the pancreatic mass.
Foregut carcinoids, similarly to pancreatic carcinoids, tend to produce an atypical carcinoid syndrome11 with increases in 5-hydroxytryptophan plasma levels, but usually not serotonin plasma levels because they lack the appropriate decarboxylase.5 Nevertheless, urinary 5-hydroxy-in-dole-acetic acid (5-HIAA) levels are markedly increased in functioning pancreatic carcinoids, suggesting a 5-hydroxytryptophan decarboxylation in the intestine and other tissues.8
From a radiological point of view, the pancreatic carcinoid tumor may be indistinguishable from pancreatic islet cell tumors, as they have some common features.16 Echography of pancreatic carcinoid reveals the presence of round or oval tumors that are generally well-defined and hypoechoic in contrast to the normal neighboring pancreatic tissue. A hyperechoic capsule may be delimited by the gland itself or may protrude from its surface. CT examination may detect well-circumscribed, homogeneous, hypodense areas that may contain calcification. Following administration of intravenous contrast material, there is a marked enhancement of the tumor, as opposed to the pancreatic ductal carcinoma.17 Angiography of the celiac axis and superior mesenteric artery may confirm the diagnosis of an endocrine tumor by its marked vascularization and may help to distinguish invasion from displacement of adjacent large vessels.5 Fine-needle aspiration biopsy with cytological examination and immunohistochemical techniques may evidence serotonin or other hormones in tumor cells and thereby enable a diagnosis of a carcinoid or a neuroendocrine pancreatic tumor.5
Although bulky tumors are frequently resectable as a curative measure, the high incidence of distant metastases precludes long-term survival for most patients, leading to an unfavorable overall prognosis.5,8–10 Pancreatic carcinoids present with a more advanced stage than other foregut carcinoids, and outcomes are less favorable than for patients with gastric and duodenal tumors.9 However, when there is no metastatic disease, surgical excision offers the best chance for recovery or long-term survival.5,8,9
The high incidence of metastases described in pancreatic carcinoids probably results from the late onset of clinical symptoms and the consequent late diagnosis, thereby allowing sufficient time for metastasis of the tumor.5,8,9 Thus, tumor stage tends to be significantly more advanced in pancreatic carcinoids, with a very strong negative impact on outcome; hence it may be said that pancreatic carcinoids are not necessarily more aggressive than other carcinoids.9 Additionally, tumor diameter does not correspond with the presence or absence of metastases.5
In cases with hepatic metastatic lesions, partial hepatic resection or intra-arterial embolization of the metastatic nodules has been attempted.18 For symptomatic treatment of inoperable tumors, subcutaneous injection of somatostatin-like substances or the administration of serotonin or histamine antagonists have been used in addition to supportive chemotherapy with streptozotocin and 5-fluor-ouracil.18
Recognition of carcinoid tumors of the pancreas is important, since this type of tumor has better a prognosis than the adenocarcinomas if detected early; additionally, effective treatment is available to combat the distressing symptoms due to the carcinoid syndrome. Accordingly, the reports of cases of primary pancreatic carcinoid should support the differential diagnosis of diarrheogenic syndrome and provide new insight into the biology of carcinoid tumors and the mechanisms of their secretagogue action in the release of amines and peptides.