




Mutations in the ARMC5 (armadillo repeat containing 5, OMIM 615549) gene, a putative tumor suppressor gene, have recently been identified as a common cause of sporadic and familial bilateral macronodular adrenal hyperplasia (BMAH). Familial BMAH is thought to be caused by two mutations, one germline and the other somatic, as suggested by the 2-hit theory. The objective is to describe a new mutation and develop its clinical characteristics and implications.
Methods, Results and ConclusionsWe present an affected family with 11 members carrying a novel mutation of the ARMC5 gene (NM_001288767.1): c.2162T>C p. (Leu721Pro). Two of the carriers developed clinical Cushing's syndrome (CS), two mild autonomous cortisol secretion (MACS) and one presented with autonomous cortisol secretion (ACS). Four patients developed other tumors, three of whom died from this cause. It is not known whether these tumors could be related to the described mutation.
Las mutaciones en el gen armadillo repeat containing 5(ARMC5), OMIM 615549, un gen supresor tumoral, se ha identificado como una causa común de hiperplasia suprarrenal esporádica y familiar. Se cree que esta enfermedad está causada por 2 mutaciones, una germinal y otra somática, basada en la teoría del doble golpe. El objetivo es describir una nueva mutación y desarrollar sus características clínicas e implicaciones.
Métodos, resultados y conclusionesPresentamos una familia afectada con 11 miembros portadores de una nueva mutación del gen ARMC5 (NM_001288767.1): c.2162T>C p. (Leu721Pro). Dos de los portadores desarrollaron síndrome de Cushing clínico, 2 secreción autónoma de cortisol leve y otro secreción autónoma de cortisol. Cuatro pacientes desarrollaron otros tumores, 3 de los cuales fallecieron por esta causa. Se desconoce si estos tumores podrían estar relacionados con la mutación descrita.
There are some genes that protect the human genome. Tumor suppressor genes act as guardians of the genome, preventing the development of neoplasia. The inactivation of these genes is a fundamental event in the pathogenesis of human cancer.1 In this sense, the armadillo repeat containing 5 (ARMC5) gene acts as a tumor suppressor gene.2–4 In vitro studies have shown that ARMC5 induces cellular apoptosis, whereas inactivation of ARMC5 abolishes this effect.5 Therefore, inactivation of ARMC5 could lead to resistance to apoptosis.
It has also been shown that germline mutations in the ARMC5 gene determine adrenal hyperplasia of adrenocortical cells with the development of nodules with hyperproduction of cortisol, which is a common cause of bilateral macronodular adrenal hyperplasia (BMAH). Theoretically, these mutations in the ARMC5 gene could also increase susceptibility to the development of other neoplastic processes, and the co-existence of other tumors in patients with BMAH has been described.6
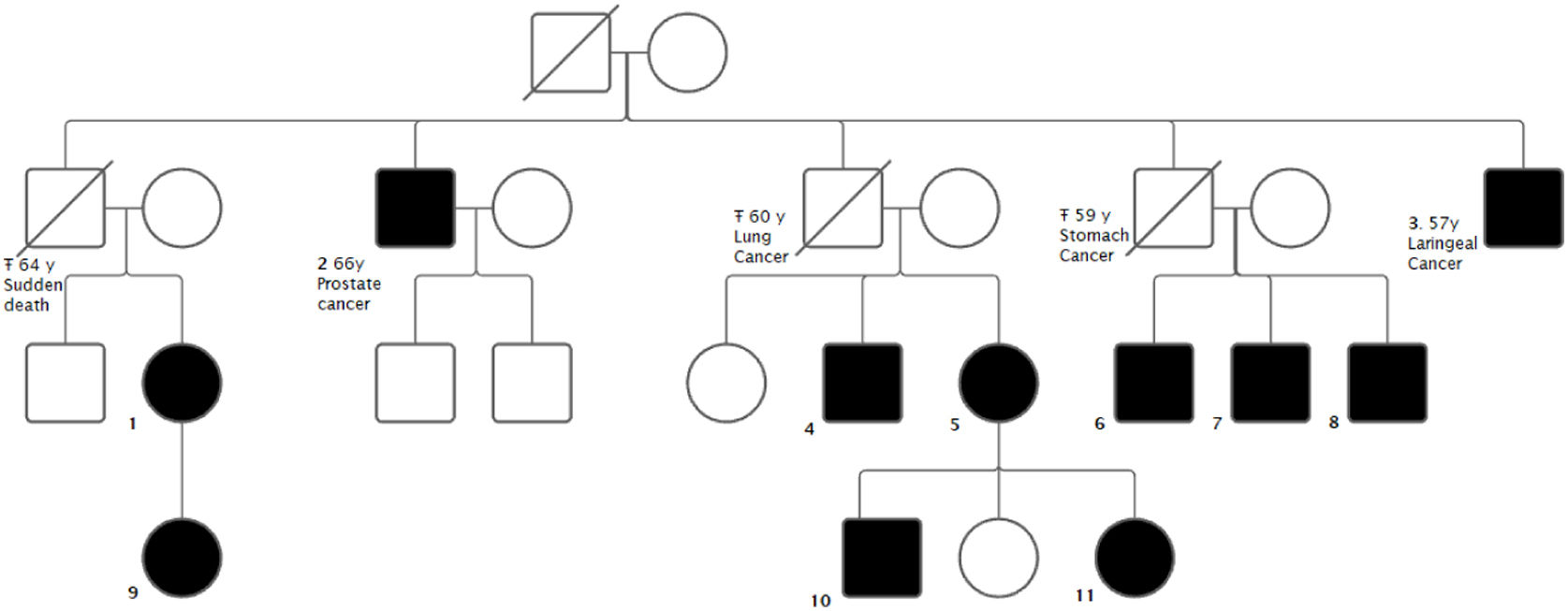
The aim of the study is to present a family with 11 carriers of a novel mutation in the ARMC5 gene. Several carriers presented with BMAH: 2 with clinical Cushing's syndrome (CS), 2 presented with mild autonomous cortisol secretion (MACS) and 1 with autonomous cortisol secretion (ACS). Four carriers presented with malignant neoplasms: one prostate cancer and three obligate carriers (not tested) due to having offspring carrying the mutation who died of malignant neoplasms - carcinoma of the larynx, carcinoma of the stomach and carcinoma of the lung.
MethodsA retrospective observational study was performed in which a family with ARMC5 gene mutation carriers was included.
The variables included were: age, sex, presence of ARMC5 gene mutation, imaging tests performed, results of cortisol suppression test with 1mg dexamethasone, surgeries performed, presence of tumors or other comorbidities and deaths.
Genetic testing was performed in all living relatives by capillary sequencing of exon 7 of the ARMC5 gene (NM_001288767.1). The adrenal function of the patients was classified according to the Criteria of the European Society of Endocrinology.7
This study was approved by the ethics committee of the Hospital Universitario Virgen del Rocío.
ResultsA family of 25 members was included.
The index case was a 48-year-old woman who was referred to our clinic for bilateral adrenal incidentalomas. She had no relevant past medical history. On initial examination, she presented with a dexamethasone suppression test (1mg) with a result in the range of mild autonomous cortisol secretion. Two years later, she developed symptoms suggestive of hypercortisolism (weight gain, asthenia, capillary fragility and hypertension). The diagnosis of CS was confirmed through pathology testing, including an overnight dexamethasone suppression test, two urinary free cortisol tests, and two nocturnal salivary cortisol tests. An I-cholesterol scintigraphy and a SPECT-CT revealed predominantly right bilateral nodular adrenocortical hyperplasia. The patient underwent total right and subtotal left adrenalectomy and remained in a situation of normocortisolism.
A 77-year-old uncle of the index case was diagnosed in another center with CS with BMAH and underwent a 2-stage adrenalectomy. Because of the suspicion of familial BMAH syndrome, a genetic study of the mutation in the ARMC5 gene was performed in both patients and a new mutation was found: (NM_001288767.1): c.2162T>C p. (Leu721Pro).
Subsequently, all living relatives underwent genetic testing (Fig. 1). Patients with the mutation underwent non-contrast CT of the adrenal glands and a dexamethasone (1mg) suppression test (Table 1). In the previously described cases, i.e. those with Cushing's syndrome, adrenal surgery was performed.

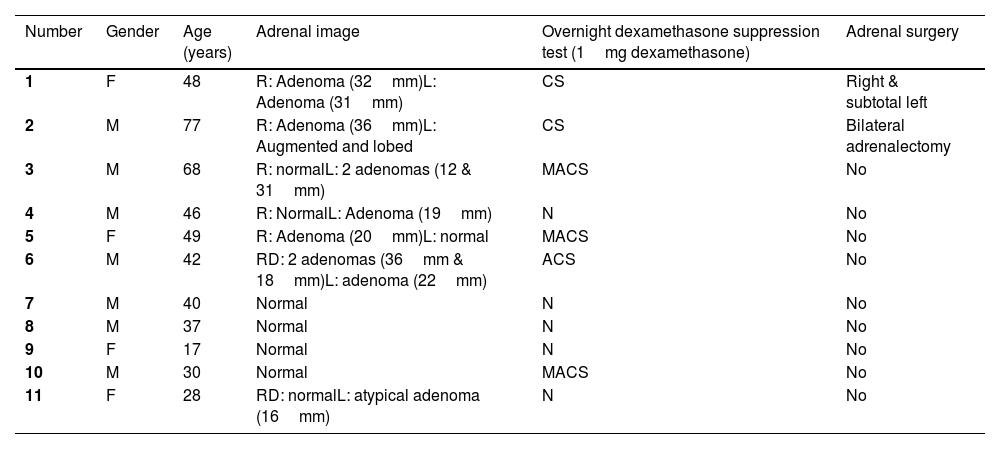
Clinical data of carriers of the new mutation in the ARMC5 gene: c.2162T>C p. (Leu721Pro).
| Number | Gender | Age (years) | Adrenal image | Overnight dexamethasone suppression test (1mg dexamethasone) | Adrenal surgery |
|---|---|---|---|---|---|
| 1 | F | 48 | R: Adenoma (32mm)L: Adenoma (31mm) | CS | Right & subtotal left |
| 2 | M | 77 | R: Adenoma (36mm)L: Augmented and lobed | CS | Bilateral adrenalectomy |
| 3 | M | 68 | R: normalL: 2 adenomas (12 & 31mm) | MACS | No |
| 4 | M | 46 | R: NormalL: Adenoma (19mm) | N | No |
| 5 | F | 49 | R: Adenoma (20mm)L: normal | MACS | No |
| 6 | M | 42 | RD: 2 adenomas (36mm & 18mm)L: adenoma (22mm) | ACS | No |
| 7 | M | 40 | Normal | N | No |
| 8 | M | 37 | Normal | N | No |
| 9 | F | 17 | Normal | N | No |
| 10 | M | 30 | Normal | MACS | No |
| 11 | F | 28 | RD: normalL: atypical adenoma (16mm) | N | No |
M=male; F=female; CS=Cushing syndrome; MACS=mild autonomous cortisol secretion; N=normal; ACS=autonomous cortisol secretion.
We consider the description of this mutation relevant due to its novelty and its possible association with the presence of tumors not previously associated with the ARMC5 gene. There are currently few published studies of individual cases or whole families with mutations in ARMC5. Our study presents one of the families with the highest number of patients with a mutation detected by family study before the onset of clinical, hormonal and/or radiological involvement.
Recently published studies on mutations in the ARMC5 gene show that adrenal nodules in patients with BMAH tend to be more aggressive and larger in size compared to patients without the genetic alteration.5 At the same time, a correlation between phenotype and genotype has been described.6 The evolution from subclinical CS stages (MACS and ACS) to a more overt form of hypercortisolism, as described in the index case, is consistent with other published studies in which there are initially no functional changes,8 with a later slow progression to subclinical and even aggressive clinical forms.
The characteristic slow development of hormonal hypersecretion means that the disease is usually diagnosed between 40 and 70 years of age, and there is a correlation between nodule size, basal cortisol levels, ACTH concentration and the age of the patient.4,6,8,9 This can be seen in our study where the younger patients, with the exception of patient 11 who already had an atypical adenoma in the left adrenal gland at the age of 18, still had normal adrenal imaging. Patients genetically affected by an ARMC5 mutation must therefore be followed up indefinitely. The underestimation of the penetrance of the disease would be in line with what has been commented.10
In the cases described above, the index case is typically between 40 and 70 years of age. The most common clinical features are usually those associated with cortisol excess: refractory or multidrug controlled hypertension,11–14 diabetes,11,12 dyslipidaemia, weight gain,11,12 osteoporosis11,15 and other physical examination signs typical of Cushing's syndrome such as moon face,12 central obesity,12 proximal muscle weakness12 or dorsal hump. In comparison, our two main cases also had typical Cushing's signs such as weight gain or capillary fragility, in addition to hypertension and weight gain. The ages were also similar to those reported elsewhere. In addition, some cases have been associated with primary hyperaldosteronism,11,16 ischemic heart disease,13 primary hyperparathyroidism,11,17 multinodular goiter,11 meningiomas11,13 or other tumors.11
In terms of radiological findings, the index case had adenomas in both glands measuring 3cm. Case 6, with autonomous cortisol secretion, also had adenomas in both glands. Case 2 had an adenoma in the right gland measuring 36mm and the contralateral gland was enlarged and lobulated. Five other patients carrying the mutation had an adenoma in only one gland, ranging from 1.6 to 31mm, with the contralateral gland normal. Four patients had normal adrenals. The radiological findings were not clearly related to the functionality of the glands, as there were patients with normal functionality and adenoma, and others with possible autonomous cortisol secretion and normal imaging. Other studies have reported images of multinodular hyperplastic glands between 1cm and 6.7cm17 as well as bilateral multinodularity with lesions up to 7.3cm.11
Regarding laboratory tests, in our study, the two patients who underwent genetic testing for a suspected ARMC5 mutation had a pathological dexamethasone suppression test with 1mg dexamethasone. Among the patients diagnosed by family study, three patients had MACS and one had ACS. The result of this test did not correlate with the age of the patient or the size of the adenomas present, as has been described in other studies.4,6,8,9 For example, this was not found in the mutant patients in the study by Giacché et al.11 where three patients had Cushing's disease and one had subclinical Cushing's disease with no clear relationship to adrenal size (the largest measured 7.3cm and the smallest 1.5cm with similar cortisol levels after suppression with 1mg dexamethasone).
Knudson's “two-hit” theory,18 described for tumor suppressor genes such as ARMC5,19 implies that the heterozygous germline mutation must be complemented by a somatic mutation to be phenotypically expressed. In the absence of a somatic mutation, the phenotypic expression is either absent or delayed because the adrenal nodules would have to reach a large size to cause CS due to inefficiency of cortisol synthesis.10 This would explain why not all members of a family who carry the mutation develop the disease. In the cases reported in this paper, the mutation could be studied in the germ line, but not in the somatic line. Other cases have been described where the somatic mutation was also not found.4,6,8,20,21
Somatic mutations in genes such as ARMC5 may contribute to the occurrence of neoplasms at other sites, although the mechanisms are not well understood.8 Meningiomas play the more important role, as their incidence is high in carriers of germline mutations in ARMC5 compared to the general population.22 Furthermore, a double hit has been demonstrated in at least one of these meningioma cases.23
Kyo C. et al.8 recently described the presence of extra-adrenal tumors (breast cancer, thyroid cancer and parathyroid cancer) in patients with germline ARMC5 mutations without somatic mutations. However, ultimately the study does not clarify the role of the ARMC5 gene alteration in non-adrenal neoplasms. In the family described in our publication, 4 carriers of the mutation presented with neoplasms. This high prevalence of neoplastic processes in first generation family members suggests a possible relationship between this novel ARMC5 mutation and the incidental occurrence of these tumors. But since there are no published data regarding a higher incidence of other malignant tumors compared to the general population, it is difficult to draw conclusions from a single family that, while they do have a higher prevalence of malignant tumors than the general population, could owe this to some other mutation or risk factor. It will be interesting to know if there are more cases of this mutation as well as the distribution of other neoplasms in these families.
ConclusionsWe have described a new mutation of the ARMC5 gene associated with BMAH and its phenotypic features. The additional peculiarity is the high incidence of other concomitant malignant neoplastic processes in its carriers. It would be interesting to establish close monitoring of carriers for early diagnosis of BMAH development and because of the possible development of other neoplasms.
Authors’ contributionsAPL and MAMC designed the study. IDLR and APG wrote the first draft of the manuscript. All authors corrected and approved the manuscript.
Financial supportThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflicts of interestThe authors declare no conflicts of interest.
