




Los síndromes paraneoplásicos endocrinos constituyen manifestaciones a distancia de algunas neoplasias. Una forma infrecuente, pero cada vez más descrita, es la osteomalacia tumoral (OT), un trastorno hipofosfatémico secundario a la pérdida renal de fosfatos inducida por la secreción tumoral del factor de crecimiento fibroblástico23 (FGF-23). Sus principales manifestaciones bioquímicas son la hipofosfatemia, la reabsorción tubular de fosfatos inadecuadamente normal o baja, los niveles bajos de calcitriol, la fosfatasa alcalina elevada y el FGF-23 sérico elevado o normal. Los tumores asociados a la OT suelen ser pequeños, benignos, de lento crecimiento, de difícil localización y con predominio en las partes blandas de los miembros. La histología más frecuente son los tumores mesenquimales fosfatúricos tipo tejido conectivo mixto. Se han propuesto varias técnicas de imagen para su identificación con resultados variables. El tratamiento de elección es la resección quirúrgica completa de la lesión. Otras alternativas terapéuticas son las sales de fósforo, el calcitriol, la octreótida, el cinacalcet y los anticuerpos monoclonales.
Endocrine paraneoplastic syndromes are distant manifestations of some tumours. An uncommon but increasingly reported form is tumour-induced osteomalacia, a hypophosphatemic disorder associated to fibroblast growth factor23 (FGF-23) secretion by tumours. The main biochemical manifestations of this disorder include hypophosphatemia, inappropriately low or normal tubular reabsorption of phosphate, low serum calcitriol levels, increased serum alkaline phosphatase levels, and elevated or normal serum FGF-23 levels. These tumours, usually small, benign, slow growing and difficult to discover, are mainly localized in soft tissues of the limbs. Histologically, phosphaturic mesenchymal tumours of the mixed connective tissue type are most common. Various imaging techniques have been suggested with variable results. Treatment of choice is total surgical resection of the tumour. Medical treatment includes oral phosphorus and calcitriol supplements, octreotide, cinacalcet, and monoclonal antibodies.
Los síndromes paraneoplásicos representan una constelación de signos y síntomas como consecuencia de los efectos a distancia de un tumor sobre diferentes órganos y sistemas. Estos efectos pueden estar mediados por moléculas con acción hormonal, factores de crecimiento, citocinas, desarrollo de autoinmunidad y otros factores desconocidos. El término ectópico significa secreción de una hormona por tejidos que fisiológicamente no lo hacen; sin embargo, las hormonas secretadas por tumores están generalmente presentes en las células precursoras no malignas, habitualmente en pequeñas cantidades. Así, la mayoría de las manifestaciones endocrinas tumorales están causadas por la secreción eutópica de hormonas por células previamente programadas para segregarlas.
La secreción hormonal inapropiada de las neoplasias se caracteriza por ser raramente suprimible; también es frecuente la generación de moléculas anómalas o incompletamente procesadas, con limitada actividad biológica y, en ocasiones, péptidos relacionados con determinadas hormonas (por ejemplo, el factor de crecimiento similar a insulina tipo II [IGF-II], péptido relacionado con la paratohormona). En la tabla 1 se enumeran las principales hormonas implicadas en síndromes paraneoplásicos.
Principales moléculas implicadas en síndromes paraneoplásicos endocrinos
| Factores de hipercalcemia |
| Péptido relacionado a hormona paratiroidea (PTHrp) |
| 1,25-dihidroxicolecalciferol (calcitriol) |
| Factor de necrosis tumoral (TNF) |
| Prostaglandinas |
| Hormona paratiroidea (PTH) |
| Corticotropina (ACTH) |
| Vasopresina |
| Gonadotropina coriónica humana (hCG) |
| Eritropoyetina |
| Calcitonina |
| Factor de crecimiento similar a insulina tipo II (IGF-II) |
| Hormona liberadora de somatotropina (GHRH) |
| Lactógeno placentario humano (hPL) |
| Hormona de crecimiento (GH) |
| Hormona liberadora de corticotropina (CRH) |
| Péptido natriurético auricular (ANP) |
| Endotelina |
| Renina |
| Hormonas gastrointestinales (somatostatina, péptido liberador de gastrina, etc.) |
| Factor de crecimiento fibroblástico 23 (FGF-23) |
La osteomalacia es una enfermedad ósea metabólica que se caracteriza por un defecto en la mineralización de la matriz ósea. En la infancia, este trastorno se denomina raquitismo y en este caso se altera, además, el cartílago de crecimiento. El proceso de mineralización requiere concentraciones de calcio y fosfato suficientes, y que la función celular y la estructura de la matriz ósea estén conservadas. Así, las 2 principales causas de osteomalacia son las alteraciones del metabolismo de la vitamina D y del fosfato. Existen otros procesos poco frecuentes que pueden interferir con la mineralización ósea, entre los que se incluyen: alteraciones de la fosfatasa alcalina, algunos fármacos y trastornos de la propia matriz ósea (tabla 2). Dentro de las osteomalacias hipofosfatémicas, una etiología poco frecuente es la osteomalacia tumoral (OT), también llamada osteomalacia oncogénica o inducida por tumores (tumor-induced osteomalacia [TIO])1,2. Se trata de un síndrome paraneoplásico causado por la pérdida renal de fósforo, descrita inicialmente por McCance en 19473, aunque su vinculación con un factor humoral se atribuye a Prader en 1959. Más adelante, se empleó el término fosfatoninas en referencia a los factores humorales fosfatúricos, y a principios de este siglo, se identificó el rol central del factor de crecimiento fibroblástico23 (FGF-23) en cuadros de osteomalacia hipofosfatémica4. En la literatura médica se han comunicado menos de 400casos, lo cual refleja su baja incidencia, las dificultades en su identificación y, probablemente, su infradiagnóstico5. La OT puede presentarse a cualquier edad, siendo más frecuente en adultos de 50-70años6.
Causas de osteomalacia
| Deficiencia o anomalías metabólicas de la vitamina D |
| Ingesta inadecuada |
| Escasa exposición a la luz solar |
| Uso de cremas con filtro de radiaciones ultravioletas (factor protección >8) |
| Hiperpigmentación cutánea |
| Edad avanzada (disminución de la síntesis cutánea de vitamina D) |
| Malabsorción (gastrectomía, bypass gástrico, celiaquía, colestiramina, colestasis crónicas, etc.) |
| Aumento del catabolismo de la vitamina D (anticonvulsivantes, tuberculostáticos, corticoides, etc.) |
| Deficiencia de 25-hidroxilación hepática (hepatopatía crónica grave) |
| Deficiencia de 1α-hidroxilación renal (fallo renal crónico, raquitismo vitamina D dependiente tipo I) |
| Pérdida renal de 25-hidroxivitamina D (síndrome nefrótico) |
| Anomalías del receptor de 1-25(OH)2D (raquitismo vitamina D dependiente tipo II) |
| Osteomalacias hipofosfatémicas |
| Raquitismo hipofosfatémico ligado al cromosoma X |
| Raquitismo hipofosfatémico autosómico (dominante, recesivo) |
| Raquitismo hipofosfatémico con hipercalciuria |
| Displasia fibrosa ósea |
| Síndrome de Fanconi (congénito, adquirido) |
| Tubulopatías adquiridas (acidosis tubular renal, metales pesados, fármacos, mieloma múltiple, etc.) |
| Tratamiento con hierro intravenoso |
| Tratamiento antirretroviral |
| Baja ingestión de fosfato asociada a la toma de antiácidos no absorbibles |
| Osteomalacia tumoral |
| Otras causas de osteomalacia |
| Ureterosigmoidostomía |
| Tratamiento farmacológico con compuestos fluorados, bisfosfonatos, aluminio |
| Hipofosfatasia congénita |
| Fibrogénesis imperfecta |
| Osteomalacia axial |
Las variantes crónicas de hipofosfatemia se asocian a manifestaciones clínicas musculares (mialgias, debilidad, miopatía proximal) y óseas (raquitismo en niños y osteomalacia en adultos). Se describen 3 mecanismos fisiopatológicos principales: la redistribución del fósforo (paso del medio interno al interior de las células), la disminución de su absorción intestinal, o el aumento de su excreción renal. Los principales reguladores del metabolismo del fósforo son la hormona paratiroidea (PTH), la 1-25 dihidroxivitamina D o calcitriol (1-25[OH]2D) y el FGF-23. Este último es normalmente expresado por los osteocitos y regula el metabolismo del fósforo y la vitamina D mediante su unión al complejo receptor Klotho-FGF7. A nivel renal, actúa disminuyendo la reabsorción tubular de fosfatos mediante la inhibición de la expresión de los cotransportadores de sodio/fosfato tipo 2a y 2c (NaPi-2a2c) e inhibiendo la actividad de la 1α-hidroxilasa renal; da lugar así a hipofosfatemia, hiperfosfaturia y niveles bajos de calcitriol. El FGF-23 está implicado en la fisiopatología de varios trastornos hipofosfatémicos. Se han identificado otros factores fosfatúricos como el factor de crecimiento fibroblástico7 (FGF-7), la matrix extracelular phosphoglycoprotein (MEPE) y la secreted frizzled related protein4 (sFRP4), pero su papel en el desarrollo de la OT aún no ha sido definido.
Manifestaciones clínicasLos síntomas y signos de la OT son similares a los de la osteomalacia hipofosfatémica familiar. Las principales manifestaciones clínicas en el adulto son: el dolor óseo, la debilidad muscular proximal y las fracturas por insuficiencia, especialmente en huesos que soportan carga como la pelvis y los miembros inferiores. Radiológicamente, se observan pseudofracturas o zonas de Looser-Milkman múltiples, una manifestación característica, aunque no específica; aparecen como bandas radiolúcidas perpendiculares a la cortical, generalmente bilaterales y simétricas, que en ocasiones progresan a fracturas completas. Sus localizaciones más frecuentes son: las costillas, las ramas pubianas, el borde externo de la escápula, el borde interno del fémur proximal y los metatarsianos. La gammagrafía ósea, más sensible que las radiografías en la localización de las pseudofracturas, muestra zonas aisladas de hipercaptación que pueden confundirse con metástasis óseas. También puede observarse un aumento generalizado de la captación isotópica (imagen de superscan), especialmente en el cráneo, la mandíbula y las articulaciones condrocostales, debido al hiperparatiroidismo secundario. El cuadro clínico de la OT es insidioso, progresivo y con síntomas inespecíficos, por lo cual suele confundirse con enfermedades reumáticas, oncológicas, psiquiátricas y otras. Esto lleva a un retraso variable en el diagnóstico correcto, que puede ser de hasta 20años. Además, la demora reportada entre el diagnóstico bioquímico de osteomalacia hipofosfatémica y la identificación del tumor varía entre 2 y 5años, ya que estos son benignos en su gran mayoría, pero de pequeño tamaño, ocultos o poco evidentes y de difícil localización5.
Hallazgos bioquímicosEl diagnóstico bioquímico se fundamenta en el hallazgo de hipofosfatemia, hiperfosfaturia, disminución de la reabsorción tubular de fosfatos (RTP), niveles bajos o inapropiadamente normales de 1-25(OH)2D sérico y niveles altos o inapropiadamente normales de FGF-23 plasmático. Bajo condiciones fisiológicas normales, el 85-95% del fósforo filtrado por el glomérulo renal es reabsorbido, en gran medida, en el túbulo proximal (∼85%), y en menor proporción, en el túbulo contorneado distal (∼15%). El rango normal de fosfaturia es amplio, pero en presencia de hipofosfatemia, una RTP inferior al 95% indica una pérdida urinaria inapropiada. Algunos autores recomiendan la determinación del umbral tubular de fosfatos ajustado por filtrado glomerular (TmP/GFR), ya que sus valores son independientes del nivel de fósforo plasmático y de la función renal, aunque varía según la edad y el sexo y no ha sido validado en poblaciones grandes8. Es posible un hiperparatiroidismo secundario como respuesta fisiológica a los niveles bajos de 1-25(OH)2D. La fosfatasa alcalina total y su fracción ósea se encuentran elevadas en grados variables por aumento de la actividad osteoblástica. La determinación del FGF-23 sérico mediante el análisis de inmunoabsorción ligada a las enzimas (ELISA) puede confirmar el diagnóstico clínico con una sensibilidad estimada del 23-86% en la OT sin tumor identificado y del 38-100% con tumor confirmado; esta variabilidad puede atribuirse a la heterogeneidad de los grupos de pacientes estudiados9,10. Sin embargo, esta técnica no está disponible de forma generalizada, y, además, se han descrito cuadros de OT con el FGF-23 sérico normal11. El diagnóstico diferencial incluye los trastornos genéticos relacionados (tabla 2), en particular algunas formas de raquitismo hipofosfatémico familiar autosómico recesivo de presentación tardía en adultos; también debe establecerse con el síndrome de Fanconi hereditario o adquirido, otras formas de tubulopatías con pérdida renal de fosfatos (metales pesados, fármacos, mieloma múltiple, etc.) y diversos trastornos que cursan con hipofosfatemia (fallo hepático, malabsorción, alcoholismo, etc.).
HistopatologíaLos tumores causantes de la OT suelen ser pequeños, benignos, de crecimiento lento y se localizan frecuentemente en las extremidades, tanto en los huesos como en los tejidos blandos; también se han encontrado en los senos paranasales, la nasofaringe, el cerebro, los ovarios, la columna y la pelvis12–14. Existen casos en los cuales no se localiza el tumor; en una serie publicada, esto ocurrió en más de un tercio de los pacientes5. Algunos autores se refieren a esta situación como síndrome similar a OT (TIO-like) o también OT secundaria, que se ha asociado a diversos tumores como sarcomas, carcinomas de próstata y mama, mieloma múltiple, leucemia linfocítica crónica y carcinomas de células pequeñas. El síndrome paraneoplásico que hoy se reconoce como OT se asocia a tumores de origen mesenquimal en general benignos, descritos anteriormente como tumores de células gigantes, fibromas osificantes, osteoblastomas, granulomas, hemangiopericitomas y otras denominaciones. Weidner y col. propusieron el término tumores mesenquimales fosfatúricos y una subdivisión en categorías: tejidos conectivos mixtos, tumores símil-osteoblastoma, tumores símil-fibroma no osificante, tumores símil-fibroma osificante y tumores metastásicos15. En conjunto, constituyen una entidad histopatológica distintiva, aunque morfológicamente heterogénea16. La primera variante es la más frecuente y comprende alrededor del 75% de los casos; en la literatura anglosajona se identifican como phosphaturic mesenchymal tumor-mixed connective tissue (PMT-MCT). Se caracterizan por presentar células gigantes similares a osteoclastos, estroma mixoide o condromixoide, actividad mitótica baja o ausente, áreas osificadas e importante vascularización, con vasos de diferente tamaño y patrón morfológico. La anterior carencia de criterios uniformes para su reconocimiento sería responsable de cierta confusión en el diagnóstico, aunque en última instancia es posible realizar una biopsia ósea sin decalcificar previamente marcada con tetraciclina, que muestra un aumento del osteoide junto a un tiempo de desfase de mineralización mayor a 100días17. Si bien en general se trata de tumores benignos, también se han reportado presentaciones malignas y con metástasis18.
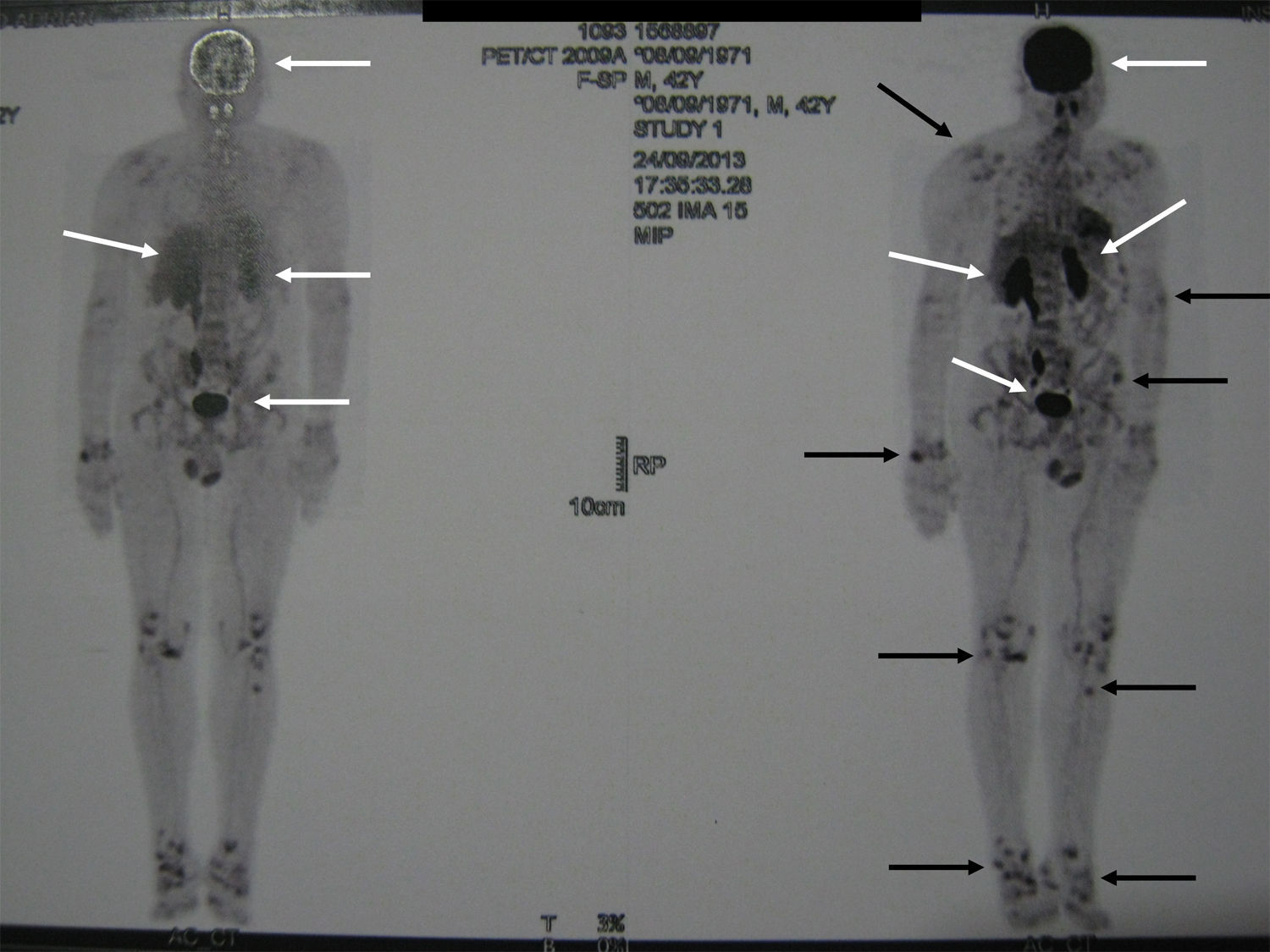
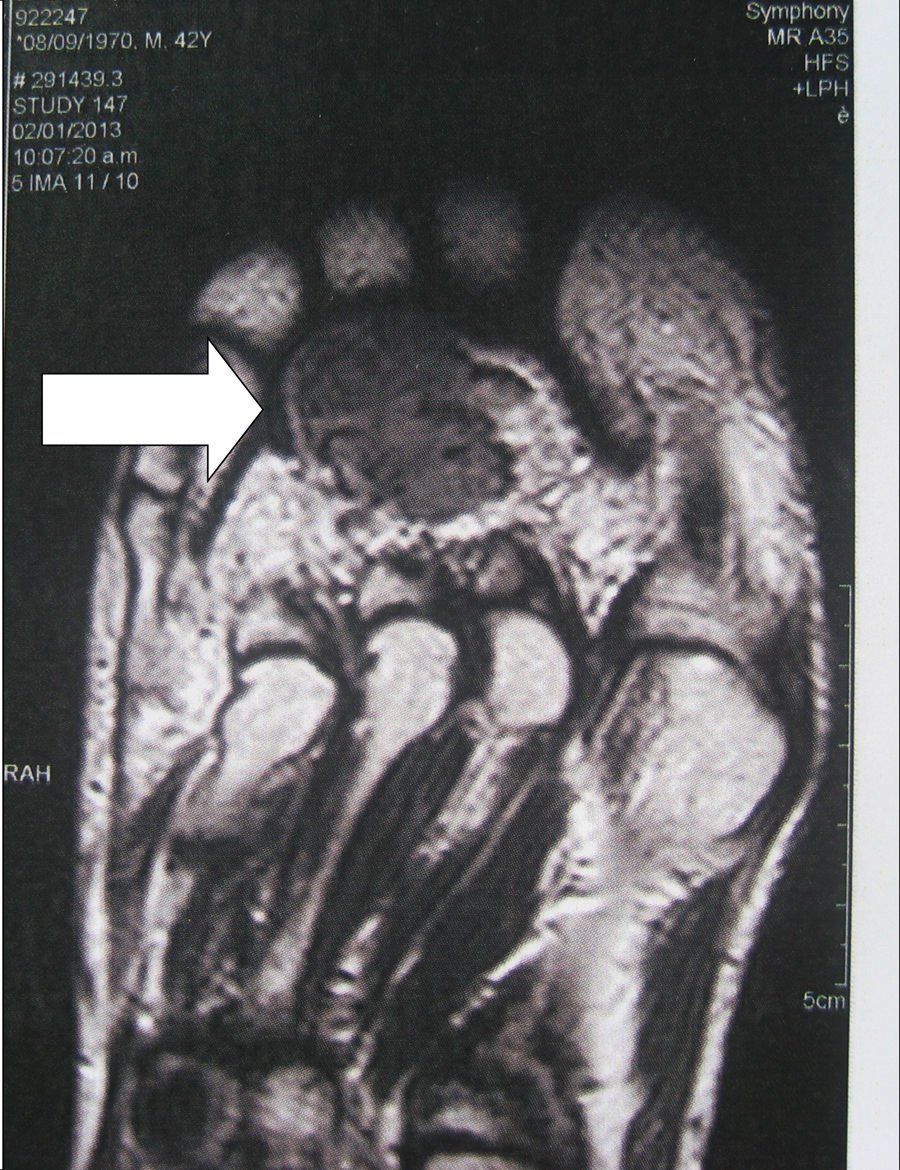
Métodos de localizaciónEs importante realizar una búsqueda exhaustiva mediante un examen físico detallado. Cuando el tumor no es evidente, se utilizan técnicas de imagen y estudios funcionales. Dadas las características de estos tumores, es frecuente que con las técnicas de imagen convencionales no se logre su detección. Como métodos de localización se han propuesto: la tomografía axial computarizada (TAC) de senos paranasales, la resonancia nuclear magnética (RNM) corporal total19, la tomografía por emisión de positrones con fluorodesoxiglucosa (FDG-PET/CT)20, la gammagrafía con octreótide marcado con 111In (octreoscan)21 y la gammagrafía con 99Tc o 201Th-sestamibi22. En los últimos años se han incorporado el Ga68-DOTANOC PET/CT (utilizando un molécula de octreótide modificada)23, o el muestreo venoso con dosaje de FGF-23 en zonas donde los estudios funcionales sugieren lesiones sospechosas22. El FDG-PET/CT es un método de alta sensibilidad, aunque con baja especificidad, sobre todo en pacientes que tienen muchas áreas de pseudofracturas, fracturas en curación o zonas líticas20 (fig. 1). La captación fisiológica en el cerebro, el hígado y el bazo puede dificultar la identificación de tumores en esas áreas y se recomiendan otros métodos de imagen para evaluar dichas localizaciones. El estudio debe incluir todo el cuerpo, ya que a veces se excluyen porciones distales de la cabeza y los miembros inferiores, donde se localizan muchos de estos tumores. La identificación de focos hipermetabólicos sospechosos requiere la confirmación anatómica con RNM o TAC (fig. 2). No obstante el avance en las técnicas descritas, en ocasiones no se localiza el tumor; en estos casos se recomienda repetir los estudios en uno o 2años2.
. Nótese la captación fisiológica en encéfalo, hígado, bazo y vías urinarias (flechas blancas).")

El tratamiento de elección consiste en la resección quirúrgica completa del tumor con amplio margen, ya que se han descrito recurrencias posquirúrgicas24. Cuando la intervención es exitosa, el cuadro clínico y bioquímico se resuelve progresivamente, aunque algunas de sus manifestaciones pueden persistir varios meses y, en general, hay secuelas óseas permanentes. Comúnmente, la fosfatemia se normaliza al cabo de unos 2-10días posteriores a la cirugía. La recurrencia tardía por metástasis es posible, aunque infrecuente; en este caso, el compromiso pulmonar ha sido el más reportado5. En tanto se concreta la cirugía, o en los casos en que no se puede realizar la exéresis completa del tumor o bien hay recurrencia posquirúrgica, se indica un tratamiento médico con suplementos orales de sales de fósforo (15-60mg/kg/día, en general 1-3g/día de fósforo elemental en 4-6tomas diarias) y de calcitriol (0,50-1,0μg/día) tomado de forma separada, con lo que se puede conseguir una mejoría clínica y bioquímica. El tratamiento debe de ser individualizado según la edad, el peso, los niveles de PTH y la función renal. Esta terapia se debe monitorizar estrechamente para ajustar la respuesta bioquímica según la tolerancia y para prevenir la hipercalcemia, la hipercalciuria, la nefrolitiasis y la nefrocalcinosis. Se han ensayado los agonistas de receptores de somatostatina (octreótide de acción prolongada por vía parenteral) con respuestas variables25. Otro posible tratamiento, basado en las interacciones entre el FGF-23 y la PTH, es el cinacalcet, un agonista del receptor sensor de calcio26. También se han comunicado casos tratados mediante ablación por radiofrecuencia e inyección intratumoral de etanol con buenos resultados27. El uso de asociaciones de quimioterapia y radioterapia han dado resultados limitados. Un enfoque novedoso es la utilización de anticuerpos monoclonales que interrumpen la interacción del FGF-23 con su receptor28.
ConclusiónLas casuística publicada en la literatura destaca las principales características de la OT: la demora diagnóstica, la difícil localización de los tumores —que son de naturaleza generalmente benigna—, el predominio en los miembros inferiores, la curación tras su extirpación completa y la posibilidad de recidivas5,29–31. La mayoría de los casos publicados a nivel internacional han aparecido en los últimos años; esto induce a pensar que se trata de una entidad probablemente subdiagnosticada previamente, siendo importante difundir su conocimiento y los criterios uniformes para su identificación. La Organización Mundial de Salud estima que hay alrededor de 7.000 enfermedades raras, poco comunes o minoritarias, que afectan al 7% de la población mundial. Este concepto engloba a un conjunto de enfermedades de baja incidencia, con dificultades diagnósticas, falta de información y conocimiento científico, limitaciones en la investigación por falta de inversiones o recursos y carencias en las terapias. La OT es una enfermedad infrecuente y plantea un verdadero desafío para el médico internista; a pesar de ello y considerando la posibilidad de curación tras la resección completa del tumor, es relevante tener en cuenta su existencia, y solicitar y evaluar los estudios diagnósticos adecuados para proceder cuanto antes a identificar y resecar el tumor fosfatúrico.
FinanciaciónLos autores no han recibido beca ni soporte financiero para la realización de este manuscrito.
Autoría/colaboradoresGuillermo Alonso y Mariela Varsavsky participaron en la concepción, diseño del manuscrito, recogida de datos, análisis e interpretación de los datos, redacción, revisión y aprobación del manuscrito remitido.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
