






Actualización en el tratamiento de la enfermedad renal crónica
More infoSon varios los factores de riesgo que pueden afectar la progresión de la enfermedad renal crónica (ERC). La hipertensión arterial, la proteinuria, la obesidad, la hipertensión intraglomerular, el tabaco y el control metabólico en la diabetes mellitus son los principales factores de riesgo de progresión modificables.
En la progresión de la ERC participan muchos procesos celulares que se originan en compartimentos específicos del riñón, compartimento vascular con nefroangioesclerosis y compartimento tubulointersticial con fibrosis y atrofia tubulointersticial, pudiendo haber imbricación entre ambos mecanismos.
Dada la implicación de tantos factores de riesgo y tantas vías patogénicas en la progresión de la ERC, la mejor esperanza para retrasar o prevenir la progresión de la ERC reside en un enfoque terapéutico combinado y multidisciplinar, basado en las evidencias existentes y que actúe sobre todos estos procesos y vías desde el punto mecanístico, y sobre un proceso global que es el riesgo cardiovascular y renal para mejorar el pronóstico de los pacientes.
Several risk factors may affect the progression of chronic kidney disease (CKD). Arterial hypertension, proteinuria, obesity, intraglomerular hypertension, smoking and metabolic control in diabetes mellitus are the main modifiable risk factors for progression.
The progression of CKD involves many cellular processes that originate in specific compartments of the kidney, the vascular compartment with nephroangiosclerosis and the tubulointerstitial compartment with fibrosis and tubulointerstitial atrophy, and there may be overlap between both mechanisms.
Given the involvement of so many risk factors and so many pathogenic pathways in the progression of CKD, the best hope for delaying or preventing the progression of CKD lies in a combined and multidisciplinary therapeutic approach, based on the existing evidence and acting on all these processes and pathways from the mechanistic point of view, and on a global process that is cardiovascular and renal risk to improve the prognosis of patients.
La enfermedad renal crónica (ERC) es un importante problema de salud pública a nivel mundial, y afecta a más del 10% de la población española. Se asocia a elevada comorbilidad, a mal pronóstico, así como a un gran consumo de recursos en el sistema sanitario.
La organización internacional KDIGO define la ERC por la presencia, durante un periodo superior a 3meses, de alteraciones de estructura o función renal con consecuencias para la salud, independientemente de la causa, y que se manifiestan por una reducción en el filtrado glomerular (<60ml/min/1,73m2) y/o por la presencia de daño renal, albuminuria, alteraciones en el sedimento, biopsia renal o pruebas de imagen1-2.
La última clasificación KDIGO CKD de 2012, junto con las guías de la S.E.N., recomiendan detallar la causa de la ERC y clasificar en seis categorías relacionadas con la tasa de filtración glomerular (G1 a G5, con G3 dividida en 3a y 3b), pero también en base a tres niveles de albuminuria (A1, A2 y A3), cada uno evaluado de acuerdo con la relación albúmina-creatinina urinaria (en mg/g o mg/mmol) en una muestra de orina aislada preferiblemente por la mañana1-2.
La ERC no solo muestra una elevada prevalencia, sino que se asocia a peores resultados en morbilidad, mortalidad y costes3-4.
A nivel mundial, alrededor del 10% al 12% de la población general tiene ERC, lo que implica que esto involucra a 850 millones de personas1,5,6. A pesar de ser en gran medida prevenible y tratable, la prevalencia de la ERC y la mortalidad por ERC están aumentando5. El estudio Global Burden of Disease proyecta que, en todo el mundo, para 2040, la ERC se convertirá en la quinta causa más común de muerte6.
La ERC puede progresar, llegando a necesitar terapia sustitutiva renal mediante diálisis o trasplante. Pero en el abordaje de este problema no solo se trata de evitar la progresión renal, sino que simultáneamente se deben utilizar tratamientos que reduzcan las complicaciones cardiovasculares, ya que los pacientes con ERC tienen seis veces más probabilidades de fallecer por enfermedad cardiovascular que de requerir tratamiento sustitutivo renal7. Por ello el abordaje de los pacientes con ERC se deberá realizar mediante la prevención y la detección de los factores de riesgo cardiovascular y progresión renal, así como evitar los fármacos y las situaciones potencialmente nefrotóxicas y la iatrogenia.
El objetivo de este artículo es analizar los factores de progresión renal, los mecanismos fisiopatológicos de progresión y su implicación con las evidencias actuales en nefroprotección.
Definición de progresión renalNo existe unanimidad en la definición de qué es progresión renal, pero dado que la tasa fisiológica de progresión renal es un descenso del filtrado glomerular estimado (FGe) de 0,7-1ml/min/1,73m2/año a partir de los 40años1-2, se considera que un paciente presenta progresión renal si presenta un descenso del FGe >5ml/min/1,73m2/año o >10ml/min/1,73m2 en 5 años. Además, también se puede considerar progresión cuando se avanza hacia una categoría de las guías KDIGO superior o más grave tanto en el FGe como en la albuminuria1-2.
La progresión renal tiene una variabilidad individual, y especialmente una variabilidad en relación con la etiología de la ERC. Las nefropatías muy proteinúricas, especialmente la glomerulonefritis y la ERC de la diabetes mellitus, tienen una mayor progresión renal. En cambio, otras etiologías tienen una progresión renal más lenta. Entre ellas mencionamos las nefropatías intersticiales (infecciones urinarias crónicas, nefrotoxicidad crónica, nefropatía por AINE, etc.) o enfermedad vascular, relativamente estable y con buen control de los factores de progresión renal, siendo clave en estos pacientes evitar la iatrogenia.
Factores de progresión renalSon muchos los factores que pueden influir en la progresión renal. Entre ellos podemos distinguir factores de susceptibilidad, como los genéticos, la raza, el sexo o el bajo peso al nacer, todos ellos no modificables, factores iniciadores de la lesión renal (mal control metabólico en la diabetes o de la presión arterial) y otros factores de progresión (hipertensión arterial, obesidad). Sobre estos últimos sí hay posibilidad de actuar (tabla 1).
Factores de progresión en la enfermedad renal crónica
| Factores de progresión | Susceptibilidad | Iniciación | Progresión |
|---|---|---|---|
| Demográficos (no modificables) | |||
| Edad avanzada | + | ||
| Sexo (varón) | + | ||
| Raza (negra, otras minorías étnicas) | + | + | |
| Masa nefronal reducida | + | + | |
| Bajo peso al nacer | + | ||
| Bajo nivel socioeconómico | + | + | |
| Hereditarios (no modificables) | |||
| Historia familiar de ERC | + | ||
| Historia familiar de enfermedad genética renal | + | ||
| Factores sistémicos | |||
| Mal control metabólico (en diabéticos) | + | + | + |
| Obesidad | + | + | + |
| Hipertensión arterial (control no óptimo) | + | + | |
| Lesión renal | |||
| Lesión renal aguda | + | + | |
| Toxinas, fármacos nefrotóxicos (sobre todo AINE) | + | + | |
| Tabaquismo | + | ||
| Problemas urológicos (infección, obstrucción) | + | + | |
| Factores relacionados con la dieta | |||
| Elevada ingesta de proteínas | + | + | |
AINE: antiinflamatorios no esteroideos; ERC: enfermedad renal crónica.
La incidencia de ERC se incrementa con la edad. En el registro americano de ERC (USRDS) la incidencia de ERC en diálisis es de 117 pacientes por millón en edades entre 18 y 44años y aumenta progresivamente hasta una incidencia de más de mil pacientes por millón en mayores de 65años8. La etiología también cambia con la edad, siendo más frecuentes la nefroangioesclerosis, la diabetes y la uropatía obstructiva en ancianos. No obstante, existe cierta controversia con la edad como factor de progresión cuando se corrige por otros factores.
GéneroSe ha descrito en estudios poblacionales como factor pronóstico independiente de padecer ERC; está claro como factor de progresión en sí mismo, habiendo varias circunstancias asociadas al sexo que son determinantes en la evolución del daño renal. No obstante, la realidad es que en todos los registros de enfermedad renal el sexo masculino representa aproximadamente al 60% de los pacientes en tratamiento renal sustitutivo9.
RazaEn Estados Unidos está demostrada una mayor incidencia en diálisis de la población afroamericana. Esta circunstancia debe atribuirse, principalmente, a la mayor prevalencia de hipertensión arterial (HTA) grave, peores circunstancias socioculturales y posibles factores genéticos10.
Bajo peso al nacerEl bajo peso al nacer está asociado a un reducido número de nefronas y al desarrollo posterior de ERC. De hecho, la pérdida adquirida de masa renal, experimental o clínica se asocia a hipertensión glomerular e hiperfiltración. Por lo tanto, es un aspecto a considerar y a incluir en la historia clínica11.
Masa nefronal reducidaHabitualmente tenemos un millón de nefronas en cada riñón, pero esta cifra puede variar entre 500.000 y 1.500.00. La masa nefronal reducida o el menor número de nefronas está en relación con el bajo peso al nacer y tener menos número de nefronas, o a la pérdida de nefronas en el tiempo por diversas patologías (evolución de la ERC, nefrotoxicidad, nefrectomía). Estas situaciones van a favorecer la hiperfiltración de las nefronas restantes y favorecer la progresión renal.
Bajo nivel socioeconómicoLos estudios epidemiológicos demuestran claramente que el bajo nivel social, cultural y económico se asocia a peor salud. La enfermedad renal no escapa a estas circunstancias12.
Factores genéticosLa realidad es que en la clínica detectamos clusters o agrupaciones familiares de pacientes con ERC sin identificar genes, síndromes concretos o alteraciones genéticas que determinen una patología. Generalmente, muchos de estos pacientes los hemos considerado como ERC de origen no filiado. Hemos avanzado mucho, pero todavía estamos lejos de conocer profundamente este aspecto en la ERC. Las nuevas técnicas de diagnóstico genético que han avanzado tanto en las últimas décadas probablemente nos van a ayudar en este aspecto.
Los factores genéticos juegan un papel importante en la ERC y su progresión. Los estudios de asociación del genoma completo (Genome-Wide Association Studies [GWAS]) han identificado ciertos loci del complejo mayor de histocompatibilidad que afectan la tasa de progresión de la ERC. Se cree que los pacientes con enfermedades monogénicas, como la poliquistosis renal autosómica dominante, que tienen el genotipo PKD1 (mutación de la policistina PKD1), tienen peor pronóstico que otros. La progresión de la ERC también puede verse influida por polimorfismos de genes que codifican mediadores putativos de la cicatrización renal, incluidos los que codifican el sistema renina-angiotensina-aldosterona.
Se ha avanzado mucho en detección del gen APOL1 implicado en la progresión renal en negros afroamericanos, o en mutaciones de los podocitos (podocitopatías) implicadas en glomeruloesclerosis segmentaria y focal progresiva, o en el estudio de genes candidatos a la diabetes y a la enfermedad renal progresiva, o mutaciones del gen de la renina o de la uromodulina, que progresivamente nos van aclarando aspectos genéticos de la ERC y sus implicaciones en la progresión13.
Factores de riesgo modificablesHipertensión arterial sistémica e hipertensión glomerularLa HTA sistémica es una causa importante, una consecuencia y una característica de presentación de la ERC. Está presente en más del 75% de los pacientes con ERC y es una de las principales causas de enfermedad renal terminal en el mundo, la segunda causa principal en Estados Unidos y en Europa después de la diabetes. Algunos estudios experimentales y epidemiológicos han demostrado que la hipertensión sostenida contribuye significativamente a la progresión de la ERC.
Existe fuerte evidencia que vincula la progresión de la ERC con la hipertensión sistémica en la enfermedad renal diabética y no diabética. La presión arterial (PA) es clave en el tratamiento de la ERC. Se cree que la transmisión de la HTA sistémica a los lechos capilares glomerulares y la hipertensión glomerular resultante contribuyen a la progresión de la glomeruloesclerosis14. Además, varios estudios de experimentación han demostrado un efecto de la HTA en el estrés mecánico en varias células glomerulares. El aumento de la presión de perfusión causa aumentos en la tensión de la pared y el volumen glomerular, lo que resulta en el estiramiento de las células glomerulares. Estas respuestas celulares al estrés mecánico conducen a la glomeruloesclerosis y a estados profibróticos14.
Los parámetros de PA que se correlacionan con la tasa de progresión de la ERC incluyen todo tipo de mediciones de la PA (ambulatoria o en consulta, diurna, nocturna, casual o mediante monitorización ambulatoria de 24horas).
Recientemente las guías KDIGO para el manejo de la HTA en pacientes con ERC han sugerido, igual que en la población general, niveles objetivo menores de 120mmHg de sistólica utilizando la medición estandarizada de la PA e individualizando según la presencia de comorbilidades15. Los beneficios obtenidos en los estudios con estas cifras de PA probablemente estén relacionados con una disminución en la presión intraglomerular.
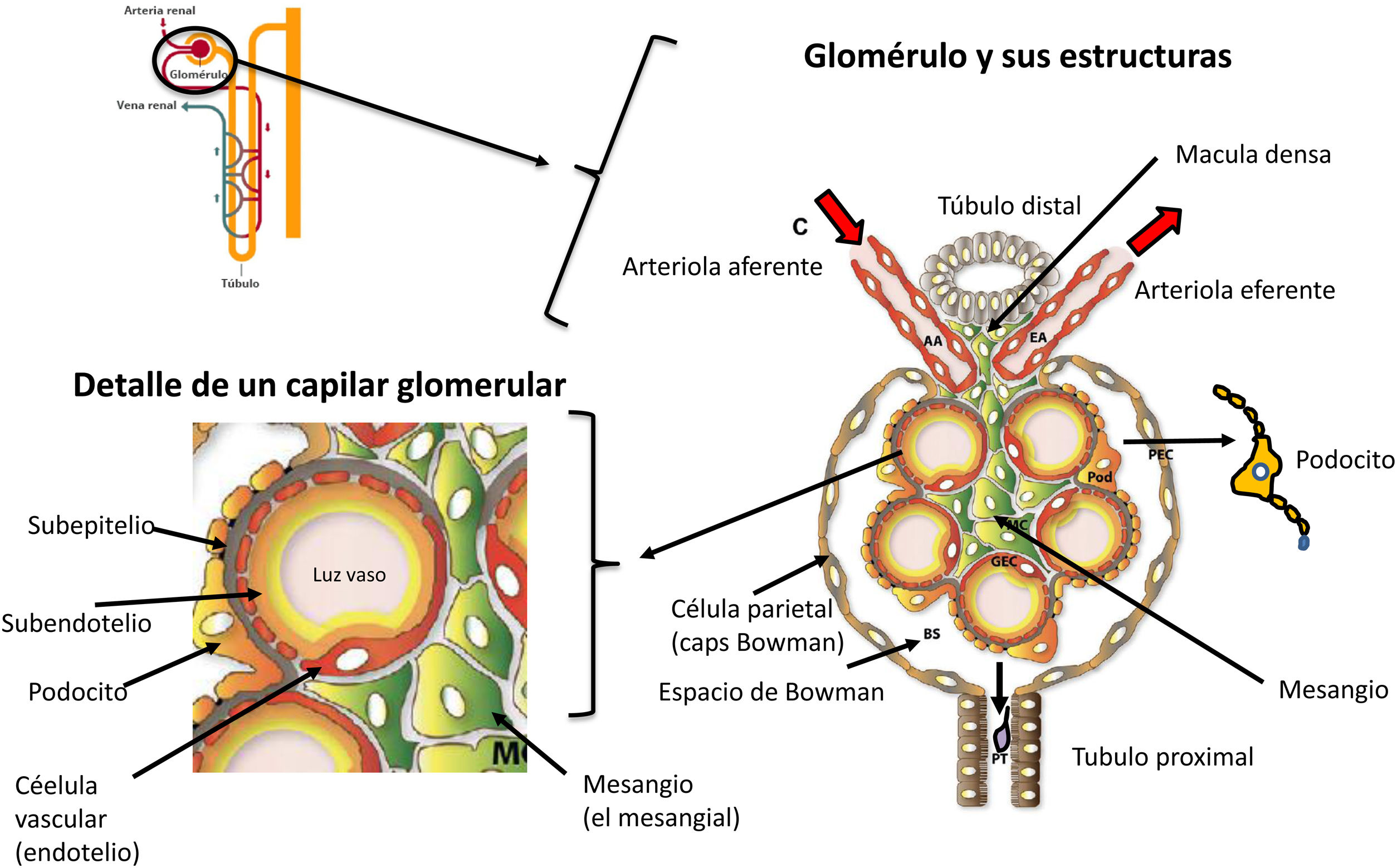
¿Cuáles son los mecanismos de beneficio renal de la reducción de la presión arterial en estos pacientes?El glomérulo tiene una estructura única, con una arteriola tanto aferente como eferente, que permite la modulación de la perfusión y la presión glomerular sin el correspondiente cambio de la PA sistémica. De manera que ante un aumento de la PA, el glomérulo desarrolla unos mecanismos de protección para evitar la transmisión de la presión hacia el interior del mismo y su microvasculatura.
En el riñón sano, estos procesos de autorregulación mantienen constantes el flujo de sangre renal y la presión capilar intraglomerular a pesar de fluctuaciones en la PA sistémica entre 80 y 170mmHg16. Esto ese consigue a través mecanismos intrínsecos (activación simpática) y extrínsecos (reflejo miógeno y feedback túbulo-glomerular):
- •
Activación simpática. Es un mecanismo intrínseco. La activación simpática (vasoconstricción de la arteriola aferente) se produce a través de la inervación de la arteriola aferente, y se pone en marcha en condiciones fisiológicas para redistribuir el flujo sanguíneo a los tejidos y también para preservar el exceso de transmisión de PA al glomérulo. Por ejemplo, al hacer deporte el flujo sanguíneo va a los músculos y al corazón. En estas situaciones se produce vasoconstricción de la arteriola aferente y disminuye el filtrado glomerular (FG) para permitir la distribución del flujo sanguíneo a áreas prioritarias. También interviene en la insuficiencia cardiaca produciendo vasoconstricción de la arteriola aferente para preservar el flujo en órganos como el corazón (coronarias) y el cerebro.
- •
Reflejo miogénico. Ante un incremento de la PA y su intento de transmisión de dicha presión al glomérulo se produce una contracción del esfínter o de la vasculatura preglomerular, que habitualmente se contrae o dilata en respuesta a incrementos o decrementos en la PA sistémica. En el riñón dañado, el reflejo miogénico está alterado y ocasiona una inadecuada autorregulación de la presión intraglomerular, de manera que la capacidad de prevenir la transmisión de la PA sistémica al interior de la circulación glomerular se pierde parcial o totalmente17-18. De esta manera, la presión intraglomerular cambia proporcionalmente con los cambios en la PA sistémica hasta tal punto que en algunos casos existe una correlación lineal entre el cambio en la PA sistémica y la presión intraglomerular. Por el contrario, si la PA media cae, se produce la dilatación de la arteriola aferente para permitir un aumento del flujo sanguíneo y el mantenimiento de una presión capilar glomerular y FGe.
- •
Feedback túbulo-glomerular. Es un mecanismo de nefroprotección que se relaciona con la función de la mácula densa y las células yuxtaglomerulares. En condiciones que aumentan el flujo sanguíneo renal (HTA, hipervolemia y aumento de la carga de sodio), y por lo tanto el filtrado glomerular, se produce una mayor llegada de cloruro de sodio a la mácula densa en el túbulo distal. Cuando las células del aparato yuxtaglomerular (situadas en la mácula densa, en contacto con el túbulo distal) detectan este aumento de la carga de cloruro sódico, median la vasoconstricción de la arteriola aferente a través de la secreción de adenosina. Como resultado hay una disminución en el flujo sanguíneo glomerular, una disminución en la presión capilar glomerular y un retorno del FG a la normalidad19.
Ello supone un importante mecanismo de nefroprotección, que en los últimos tiempos se ha conocido mejor por ser uno de los principales mecanismos de protección hemodinámica de los inhibidores de cotransportador sodio-glucosa tipo2 (iSGLT2). Tras la administración de los iSGLT2 existe una menor reabsorción de sodio en el túbulo proximal, llegando una mayor carga de sodio a la mácula densa y reestableciendo este feedback túbulo-glomerular, induciendo una vasomodulación o vasoconstricción de la arteriola aferente que disminuye la presión dentro del glomérulo y contribuye mediante mecanismo hemodinámico a la nefroprotección.
Por el contrario, cuando hay una hipotensión o disminución en el flujo sanguíneo glomerular, hay una disminución en el suministro de cloruro de sodio a las células de la mácula densa. Para mantener el FG constante se produce una dilatación de la arteriola aferente, un aumento del flujo sanguíneo glomerular, un aumento de presión capilar glomerular y un retorno del FG a la normalidad. Este mecanismo funciona más lentamente que el mecanismo miogénico, pero es crítico para el mantenimiento del FG en condiciones que provocan cambios en la presión intraarteriolar.
Numerosos estudios en pacientes con enfermedades renales diabéticas y no diabéticas confirman que la proteinuria marcada (o albuminuria gradoA3) se asocia con una tasa más rápida de progresión de la ERC20.
Es más probable que la albuminuria o la proteinuria sean un marcador de lesión glomerular o de fibrosis tubulointersticial, que una causa de la ERC progresiva porque reflejan la gravedad del daño renal subyacente. La proteinuria es el resultado de la permselectividad alterada de la barrera de filtración glomerular, causada por factores hemodinámicos y no hemodinámicos21.
En condiciones normales la barrera capilar de filtración glomerular está intacta e impide el paso de proteínas al espacio urinario. En condiciones patológicas se rompe este equilibrio y aparece una cantidad variable de albúmina y otras proteínas en el túbulo renal. En modelos experimentales se ha demostrado que la reabsorción activa por parte del túbulo de estas proteínas filtradas por el glomérulo induce lesiones histológicas en lisosomas con rotura lisosomal. A medida que se va reabsorbiendo un mayor grado de proteínas, y de forma continua, el fenotipo de las células epiteliales tubulares va cambiando a tipo mesenquimal con un infiltrado celular intersticial que es mayor cuanto mayor sea el grado de proteinuria, apareciendo finalmente zonas de fibrosis intensa. Se ha comprobado que las células del epitelio tubular tienen receptores específicos para muchas sustancias proinflamatorias y vasoactivas que pasan a la luz tubular en situaciones de proteinuria. Igualmente se ha demostrado que la presencia de proteinuria estimula la síntesis de numerosas citocinas, compuestos vasoactivos y factores de crecimiento que suponen un estímulo para la fibrosis renal22.
En la mayoría de los estudios, el mayor grado de proteinuria o la escasa respuesta de la misma a las terapias aplicadas ha sido el más potente predictor de la evolución de la enfermedad renal tanto diabética como no diabética. Por ello, uno de los objetivos principales de la nefroprotección se basa en la reducción de la albuminuria/proteinuria.
Sistema renina-angiotensina-aldosteronaEn la ERC, el vínculo entre la hipertensión arterial sistémica, la proteinuria, la albuminuria y la enfermedad cardiovascular probablemente está mediado por cambios en el sistema renina-angiotensina-aldosterona (SRAA).
Los estudios experimentales y clínicos han implicado al SRAA en la patogenia de la hipertensión, la proteinuria y la fibrosis renal a lo largo del curso de la ERC. En consecuencia, las intervenciones dirigidas a la inhibición del SRAA han resultado eficaces para retardar la progresión de la ERC20,23.
La angiotensina II es el principal efector del SRAA y ejerce su efecto vasoconstrictor predominantemente sobre las arteriolas postglomerulares, aumentando así la presión hidrostática glomerular y la ultrafiltración de proteínas plasmáticas, efectos que pueden contribuir a la aparición y a la progresión del daño renal crónico. La angiotensinaII también puede contribuir directamente a acelerar el daño renal al mantener el crecimiento celular, la inflamación y la fibrosis. Las intervenciones que inhiben la actividad del SRAA son renoprotectoras y pueden retrasar o incluso detener la progresión de las nefropatías crónicas. Estos datos han motivado que el tratamiento con bloqueadores del SRAA sea casi universal de los pacientes con ERC proteinúrica y/o diabética20,23.
Los bloqueadores de SRAA actúan dilatando la arteriola eferente y disminuyendo así la presión dentro del glomérulo. Pero además de disminuir la hiperactividad del SRAA, este grupo farmacológico actúa reduciendo la proteinuria y otras acciones mediadas por la angiotensinaII así como las fuerzas de estrés endotelial, previniendo la fibrosis intersticial24.
La combinación de IECA y antagonistas de los receptores AT1 (ARAII) podría tener una ventaja teórica, al permitir un mayor bloqueo de las acciones de angiotensinaII mientras se mantiene la disponibilidad local preferencial del receptor AT2. Pero en animales de experimentación no resultó en un beneficio adicional en la glomeruloesclerosis comparado con terapia de un solo fármaco con control similar de la PA. Y además, en ensayos clínicos dicha combinación utilizada en pacientes de elevado riesgo vascular se ha asociado a elevado riesgo de hipercalemia y a mayor mortalidad25. Por ello esta estrategia no se recomienda en las guías1,2,26.
ObesidadLa obesidad y el sobrepeso son factores conocidos de progresión renal, aunque la mayoría de los estudios sobre el impacto nefroprotector de la pérdida de peso son retrospectivos o prospectivos, pero con solo estudios observacionales de un solo brazo. En pacientes con obesidad (con y sin diabetes), la pérdida de peso induce una reducción significativa de la proteinuria27.
Los factores a través de los cuales se establece una posible relación entre la obesidad y la progresión renal son factores hemodinámicos (hiperfiltración, aumento de lesión de los podocitos, glomerulomegalia, aumento en la reabsorción de sodio y en la actividad simpática, y aumento de la actividad del SRAA), factores metabólicos (aumento de la resistencia a la insulina, inflamación, estrés oxidativo, disregulación de adipocinas y alteración del metabolismo lipídico) y lipotoxicidad (excesiva acumulación de grasa perirrenal, disfunción mitocondrial y aumento de la toxicidad tubular de los ácidos grasos libres)28.
TabacoEl tabaquismo es un reconocido factor de riesgo cardiovascular, y se propone como factor independiente de riesgo renal, aunque sus mecanismos no están establecidos. El tabaco causa aumento de la PA y vasoconstricción a nivel hemodinámico renal con disfunción endotelial (disminución de la disponibilidad de óxido nítrico y vasodilatación dependiente de las células endoteliales e hiperplasia de las células de la íntima). Además, se ha asociado a incremento de la producción de citocinas y marcadores inflamatorios. Por otra parte, el tabaco contiene glicotoxinas, que pueden inducir rápidamente la formación de productos finales de glicación avanzada tanto in vivo como in vitro, que aumentan la permeabilidad vascular y promueven los cambios vasculares patológicos de la enfermedad renal. Y finalmente, fumar se ha relacionado con la resistencia a la insulina en sujetos diabéticos como no diabéticos, siendo este un predictor independiente de ERC y albuminuria29-30.
En estudios clínicos se ha asociado a aumento en la proteinuria y a mayor progresión renal29. Recientes metaanálisis han demostrado una clara relación entre tabaquismo y progresión renal29-30. Debe considerarse uno de los más importantes factores de riesgo remediables, y por ello la abstinencia al tabaco es una recomendación prioritaria en la ERC.
LípidosEs bien conocido que la dislipemia conlleva un efecto adverso sobre el árbol vascular en general. Las alteraciones lipídicas se asocian a modulación de la esclerosis glomerular en ratas. Además, se ha detectado progresión de las lesiones renales cuando se añade un exceso de colesterol en las dietas31. A pesar de estos datos y de que varios estudios observacionales han mostrado una asociación entre los niveles de colesterol LDL y una mayor progresión renal, no se ha podido demostrar este beneficio en la progresión renal en estudios aleatorizados32. En cualquier caso, la evaluación y la intervención terapéutica para el control de la dislipemia en el paciente renal son preceptivas, dado el elevado riesgo cardiovascular que presenta el paciente con ERC, y las estatinas deben formar parte de la terapia de abordaje holístico33.
NefrotoxicidadLa nefrotoxicidad es uno de los factores de iniciación y progresión de la ERC. Los pacientes pueden presentar ERC sin que se conozca en muchos casos, y en ocasiones no se produce un ajuste de la dosis. Especialmente en situaciones de la ERC oculta, cuando la creatinina es normal, pero especialmente en ancianos, hay un FG reducido. En otras ocasiones se produce daño renal agudo que puede favorecer el daño renal crónico. En estos casos la nefrotoxicidad viene condicionada por la toma continuada de fármacos, como los antiinflamatorios no esteroideos (AINE), favorecidos por situaciones hemodinámicas agudas en las que tienen un papel importante los diuréticos o los bloqueadores del SRAA, sustancias potencialmente tóxicas o episodios de lesión renal aguda, que disminuyen el FG y aumentan el riesgo de lesión renal en un futuro2.
El riñón es particularmente vulnerable a la acción de fármacos y toxinas, ya que es el órgano que recibe mayor irrigación por gramo de tejido y es la principal vía de eliminación de fármacos y de sus metabolitos, siendo susceptible a la nefrotoxicidad de los fármacos o de combinaciones de los mismos, especialmente cuando se dan ciertas situaciones hemodinámicas, especialmente de hipoperfusión renal.
Los mecanismos de nefrotoxicidad actúan sobre el glomérulo o sobre los túbulos. Las nefrotoxinas actúan sobre los vasos sanguíneos renales produciendo una disminución del flujo sanguíneo renal. Por otra parte, actúan sobre los túbulos produciendo necrosis celular y obstrucción tubular, favoreciendo el backleak o escape de líquido desde la luz tubular. Todo ello produce como resultado una reducción en el FG. En estas circunstancias, una disminución del FG va a favorecer la isquemia tubular y consecuentemente la nefrotoxicidad por acúmulo de ciertos fármacos potencialmente nefrotóxicos cuando se reduce el FG (por ejemplo, el litio). En otras ocasiones, como en el caso de los contrastes, se produce toxicidad medular con isquemia de esta por vasoconstricción de la arteriola aferente con hipoxia de las áreas renales peor perfundidas, como es la médula renal. Además de estos mecanismos de toxicidad renal indirecta, algunas sustancias tienen un efecto directo tóxico tubular (tenofovir, aminoglucósidos) o por precipitación del fármaco en los túbulos (aciclovir)34.
Otros factoresOtros factores se han asociado también a progresión renal, pero precisan más estudios para demostrar evidencias de su implicación, tales como el estado de la microbiota, el síndrome metabólico, la resistencia a la insulina, la hiperuricemia, la acidosis metabólica, la anemia, el abuso de alcohol y los remedios a base de hierbas, plomo y otros metales pesados.
Mecanismos de progresión de la enfermedad renal crónicaUna vez conocidos los factores de riesgo para la progresión de la ERC analizaremos a través de qué mecanismos celulares y moleculares se produce la progresión de la ERC que lleva a la insuficiencia renal terminal.
En la figura 1 se muestran las estructuras de la nefrona de forma esquemática para poder entender los cambios que se producen mediante los mecanismos de progresión renal. De una manera esquemática podemos definir que el riñón tiene dos compartimentos: compartimento vascular o glomerular y compartimento túbulo-intersticial, y ambos se encargan de realizar las funciones del riñón (fig. 2). Estas funciones incluyen los mecanismos hemodinámicos de regulación vascular, flujo renal e hipertensión arterial, regulación del metabolismo hidroelectrolítico y de iones divalentes (Ca, PO4, Mg), regulación del equilibrio ácido-base, excreción de productos nitrogenados, reabsorción de sustancias a nivel tubular, síntesis de calcitriol, eritropoyetina, renina y catabolismo de insulina y PTH.

. Se muestra un ejemplo en la parte superior derecha de la imagen, extrapolable a toda la muestra. El espacio que ocupan todos los túbulos es el compartimento túbulo-intersticial.")
Corte histológico renal con hematoxilina-eosina que muestra glomérulos y túbulos en un riñón normal. Esquema de los compartimentos de la nefrona. Los glomérulos se muestran rodeados de un círculo negro. Los glomérulos constituyen el compartimento vascular de la nefrona. En el riñón normal el espacio que hay entre los glomérulos está constituido por túbulos que no dejan espacio entre ellos (espacio virtual). Se muestra un ejemplo en la parte superior derecha de la imagen, extrapolable a toda la muestra. El espacio que ocupan todos los túbulos es el compartimento túbulo-intersticial.
La lesión del compartimento vascular o glomerular por progresión renal dará lugar a inflamación, mostrando histológicamente adherencias extracapilares, fibrosis y esclerosis glomerular con obliteración del ovillo vascular, reducción progresiva de la superficie de filtración glomerular y pérdida de nefronas22,35.
La lesión intersticial se inicia mediante inflamación, aumento de producción de angiotensinaII y desconexión del feedback túbulo-glomerular, mostrando en la histología un infiltrado celular mononuclear y aumento de la matriz extracelular, y la vasculatura renal muestra esclerosis y pérdida de capilares en el intersticio cortical con isquemia pericapilar tubular, finalizando con fibrosis y atrofia tubular.
El resultado de ambas lesiones es la esclerosis de las nefronas y la insuficiencia renal terminal22,35.
Mecanismos de lesión glomerular. GlomeruloesclerosisLas enfermedades glomerulares involucran procesos en área endocapilar, extracapilar o mixtos. Los procesos confinados al compartimento endocapilar generalmente no se consideran de gran relevancia en la progresión de la ERC progresiva. Por el contrario, las alteraciones en el compartimento extracapilar, en particular las alteraciones que involucran a los podocitos, preparan el escenario para la glomeruloesclerosis progresiva, la inflamación tubulointersticial, la fibrosis y la atrofia tubular. Los podocitos son las células epiteliales de soporte a nivel renal que rodean al capilar y forman parte de la barrera de filtración glomerular interdigitándose entre ellos para retener proteínas y evitar su paso a la orina. Estos procesos extracapilares que vamos a describir a continuación pueden ocurrir de novo o como extensión de procesos patobiológicos que residían inicialmente en el compartimento endocapilar22.
La glomeruloesclerosis se caracteriza por un aumento en la acumulación de matriz mesangial y obliteración de los capilares glomerulares. El mecanismo patogénico subyacente de la glomeruloesclerosis es aún más complicado y se puede dividir en varias fases distintivas (fig. 2):
- •
Fase de inflamación. En primer lugar, bajo la influencia de varios factores de riesgo como HTA, dislipidemia y/o depósito de inmunocomplejos, las células residentes en los glomérulos se lesionan y activan, siguiéndose de una liberación de múltiples citocinas y quimiocinas, que atraen a los monocitos, a los linfocitos, a los neutrófilos y a otros tipos de células inflamatorias, que migran y se acumulan en el sitio de la lesión.
- •
Fase de proliferación. En segundo lugar, estas células infiltrantes acompañadas de células intrínsecas producen además múltiples citocinas y factores de crecimiento, incluida la angiotensinaII, el factor de crecimiento transformante β1 (TGF-β1), el factor de crecimiento derivado de plaquetas (PDGF), el factor de crecimiento de fibroblastos (FGF), el factor de necrosis tumoral (TNF-α) e interferón gamma (IFN-γ) para inducir el colapso y la obliteración capilar, la pérdida de podocitos y la activación de las células del epitelio parietal de la cápsula de Bowman.
- •
Fase de fibrosis. El proceso final es la fibrogénesis, que incluye la producción de nuevos componentes de matriz extracelular para reemplazar el tejido dañado, lo que proporciona un andamiaje para el cierre, la remodelación y la reparación de heridas. Por lo tanto, la glomeruloesclerosis refleja principalmente la lesión, la proliferación y la producción de matriz de los podocitos por parte de las células mesangiales, así como el daño y la disfunción endoteliales22
Todo este mecanismo afecta a las siguientes áreas de la nefrona:
- •
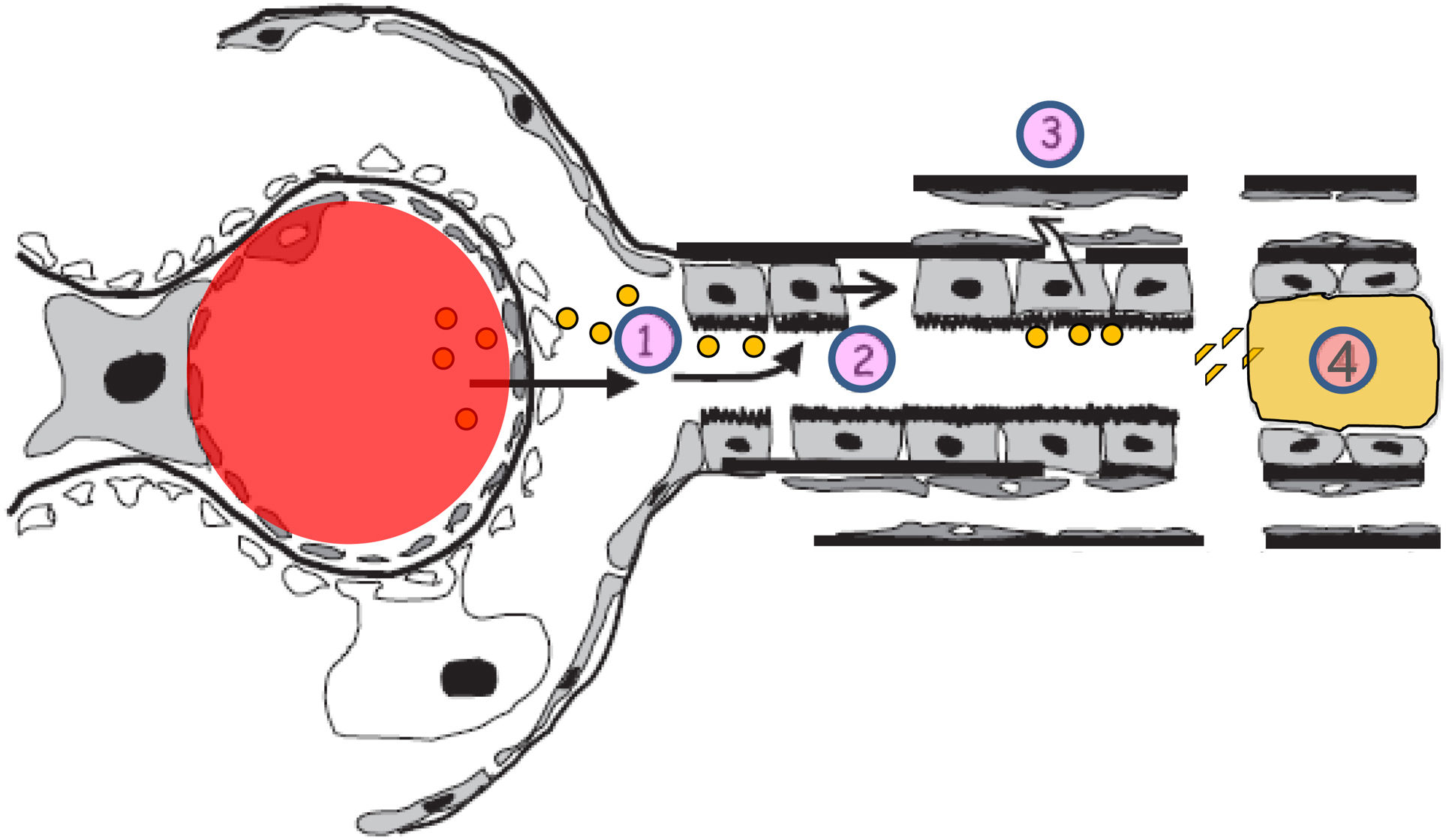
Lesión de los podocitos. El podocito es uno de los tipos de células más importantes para la barrera de filtración glomerular. El podocito y su diafragma de hendidura son la estructura principal de la barrera de filtración glomerular.Los podocitos estabilizan la barrera de filtración glomerular a través de la producción de moléculas que constituyen la membrana basal glomerular, del mantenimiento de la estructura de la membrana o diafragma de filtración y mantienen viable la célula endotelial.Los podocitos lesionados por diversos factores de riesgo (ya sea diabetes, hipertensión arterial o glomerulonefritis) sufren un desprendimiento y apoptosis, que es una lesión irreversible y que provoca una pérdida permanente. Debido a que los podocitos son células con diferenciación terminal, no pueden proliferar. El área de la membrana basal glomerular (MBG) está inicialmente cubierta por el podocito, pero al lesionarse esta queda expuesta. En estas áreas de la membrana basal glomerular expuestas o «desnudas de podocitos» se pueden adherir células epiteliales parietales. Este escenario conduce a la formación de adherencias o sinequias entre la membrana basal glomerular y las células parietales, que se reconocen como algunos de los primeros signos de glomeruloesclerosis segmentaria (fig. 3).La extensión de las sinequias conduce a la pérdida de proteínas al espacio de Bowman, y en primer lugar a la aparición de albuminuria/proteinuria, y al colapso y esclerosis del ovillo capilar asociado con pérdida de células endoteliales, lo que se asocia a la formación de semilunas y esclerosis. Estas proteínas derivadas de la filtración mal dirigida quedan atrapadas dentro del espacio de Bowman, lo que finalmente provoca la progresión a la esclerosis global y la obsolescencia del ovillo glomerular36.Además, se produce una lesión de la barrera diafragmática de la membrana basal glomerular, con un mayor borramiento de los podocitos, una mayor lesión estructural en la membrana basal y un mayor grado de proteinuria. Esto va seguido de cambios en el citoesqueleto de la MBG, con pérdida y apoptosis de podocitos35-37 (fig. 3).
- •
Expansión mesangial. Las células mesangiales son células con función de sostén entre las asas capilares glomerulares, que son elementos contráctiles y tienen un papel activo en la regulación de la filtración glomerular. Estas células responden a varias señales para contraerse y relajarse, principalmente a la angiotensinaII, gracias a lo cual regulan el flujo en los capilares intraglomerulares y con ello la filtración glomerular. Ante la presencia de inmunocomplejos circulantes, como por ejemplo en las glomerulonefritis, o por efecto de otras sustancias metabólicas o vasopresoras, las células mesangiales se activan con producción y activación del complemento y otros mediadores inflamatorios, como los prostanoides, el factor activador de plaquetas (PAF), las especies reactivas de oxígeno, las citocinas como IL-6, TNF-α, CSF-1 y las quimiocinas. Los inmunocomplejos, como los que implican la IgA, se depositan en el mesangio con activación simultánea del complemento. En respuesta a estos inmunocomplejos aberrantes, las células mesangiales se activan y producen varios mediadores de la inflamación, como quimiocinas, citocinas y factores de crecimiento, como las células mesangiales bFGF, PDGF y TGF-β. Esto da lugar a la proliferación de células mesangiales y a la expansión de la matriz mesangial, que son las primeras fases de la progresión en distintas patologías, como en las glomerulonefritis o en la ERC de la diabetes. Estas lesiones producen un círculo vicioso nocivo de entrada de macromoléculas en el mesangio que aceleran aún más la producción de mediadores inflamatorios, cambios en la permeabilidad y transmisión de lesión a los podocitos colindantes y aparición de proteinuria. En la diabetes mellitus esa activación también se produce por los productos de glicación avanzada. La progresión de estas lesiones en la diabetes mellitus da lugar a los nódulos de Kimmestiel-Wilson22,35,36.

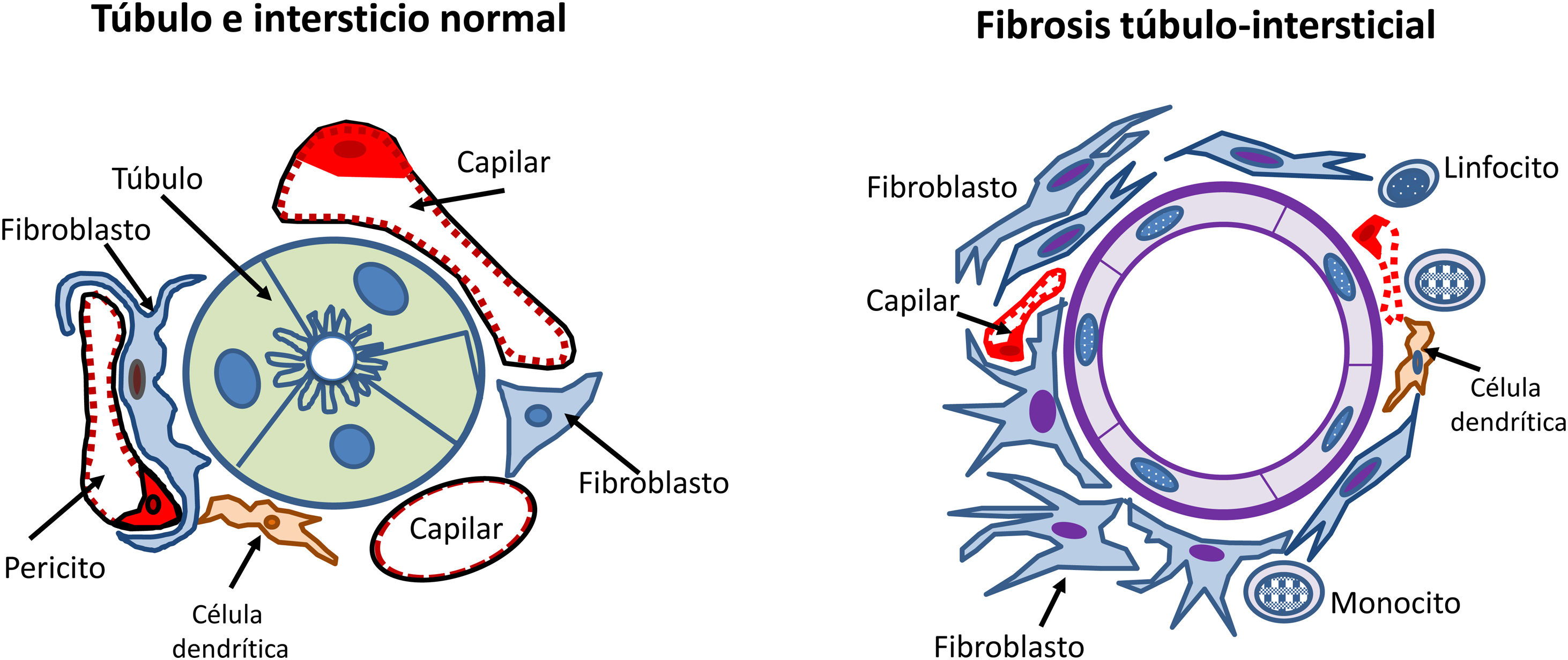
Los mecanismos de lesión tubulointersticial han ido ganando la atención en los últimos años, ya que se ha observado que la severidad de los cambios tubulointersticiales en la biopsia renal se correlaciona mejor con el pronóstico renal que las alteraciones histológicas glomerulares detectadas en la nefroangioesclerosis35.
Las células tubulares renales tienen la capacidad de actuar como células presentadoras de antígenos y expresar así moléculas de adhesión y mediadores inflamatorios, citocinas y factores de crecimiento. También responden a angiotensinaII y a otros estímulos aumentando la producción de matriz extracelular. Todos estos estímulos producen una transformación de las células tubulares con activación de los fibroblastos del intersticio renal que proliferan e invaden los espacios peritubulares y periglomerulares induciendo mayor producción de matriz extracelular que se acumula en el intersticio renal con la transformación y la proliferación de miofibroblastos. Este proceso es ayudado e instigado por células inflamatorias, principalmente monocitos y células inmunitarias residentes, incluidas las células dendríticas (fig. 4).
. En la parte inferior se muestra el efecto de la toxicidad tubular de las proteínas filtradas por el glomérulo mediante los siguientes pasos: 1.Aparición de albuminuria/proteinuria. 2.Activación de las células tubulares para sintetizar mediadores inflamatorios. 3.El daño de la membrana basal tubular facilita el paso de mediadores inflamatorios hacia el intersticio y los capilares peritubulares. 4.En la nefrona distal los cilindros de proteínas pueden obstruir el flujo urinario, además de producir daño inflamatorio.")
Cambios en el espacio túbulo-intersticial en el proceso de progresión de la enfermedad renal crónica. En la parte superior izquierda se muestran el túbulo y el intersticio normales. En la parte superior derecha se muestran los cambios relacionados con la progresión renal (inflamación, transformación de células tubulares, proliferación de matriz extracelular, activación de los fibroblastos, disminución de pericitos y densidad capilar y fibrosis). En la parte inferior se muestra el efecto de la toxicidad tubular de las proteínas filtradas por el glomérulo mediante los siguientes pasos: 1.Aparición de albuminuria/proteinuria. 2.Activación de las células tubulares para sintetizar mediadores inflamatorios. 3.El daño de la membrana basal tubular facilita el paso de mediadores inflamatorios hacia el intersticio y los capilares peritubulares. 4.En la nefrona distal los cilindros de proteínas pueden obstruir el flujo urinario, además de producir daño inflamatorio.
Si esta inflamación intersticial y la proliferación de la matriz extracelular continúan sin control, inevitablemente se produce fibrosis.
Por otra parte, y para perpetuar la lesión, ante la presencia de proteinuria, las células tubulares responden mediante la producción de mediadores inflamatorios y matriz extracelular: síntesis de quimiocinas (MCP-1, RANTES) que reclutan monocitos, célulasT e interleucinas que atraen neutrófilos y moléculas promotoras de fibrosis. El daño a la membrana basal tubular facilita el paso de productos derivados de los túbulos hacia el intersticio y los espacios de los capilares peritubulares35,37. Este mecanismo puede considerarse como un link entre lesión glomerular, la proteinuria y la fibrosis tubulointersticial, produciéndose el círculo vicioso de la progresión renal (fig. 4).
Como consecuencia de estos mecanismos se produce finalmente la fibrosis intersticial, que junto con la esclerosis glomerular derivada de los mecanismos de glomeruloesclerosis antes mencionados produce la disminución del FG, que finalmente conduce a la insuficiencia renal terminal.
Estrategias para la nefroprotección y mecanismos de beneficioAunque hasta ahora han sido escasas las herramientas disponibles para la nefroprotección, en los últimos años nuevos ensayos clínicos y terapias farmacológicas se han añadido al arsenal terapéutico de la nefroprotección. En base a los mecanismos fisiopatológicos anteriormente citados, a continuación se describen las estrategias de nefroprotección a seguir en los pacientes con ERC o con riesgo de aparición de la misma, estrategias basadas en evidencias1-2.
- •
Control de factores de riesgo cardiovascular. Incluye el control óptimo de la PA, con cifras de PA sistólica de 120mmHg si se tolera, uso de estatinas en pacientes con ERC para conseguir los niveles de colesterol LDL aconsejados por las guías1-2 y medidas generales de estilo de vida (evitar el tabaquismo, dieta sana con poca sal, ejercicio físico).
- •
Maximización del bloqueo del SRAA, utilizando a ser posible dosis plena y dieta hiposódica. Si se precisa, se puede maximizar el efecto asociando diuréticos.
- •
Optimizar el control glucémico en diabéticos (para intentar conseguir HbA1c <6,5% sin hipoglucemias). En diabéticos, utilización de hipoglucemiantes que hayan demostrado beneficio cardiovascular y renal (iSGLT2 y/o AR GLP1).
- •
Administración de iSGLT2 para el tratamiento de la ERC diabética y no diabética. En este momento solo está aprobada dapagliflozina para el tratamiento de la ERC no diabética.
- •
Finerenona en pacientes con ERC y diabetes mellitus tipo2 si no hay contraindicación por la cifra de potasio y FGe (finerenona no está actualmente comercializado en España).
- •
Evitar la dieta hiperproteica en la ERC.
- •
Prevención de la nefrotoxicidad. Evitar nefrotóxicos, minimizar a ser posible el uso de contrastes yodados intravenosos, ajustar fármacos a la función renal y detección de situaciones de deterioro renal de carácter funcional o hemodinámico.
- •
Control de complicaciones de la ERC (anemia, déficit de hierro, hiperparatiroidismo secundario, acidosis metabólica).
Este artículo forma parte del suplemento titulado «Actualización en el tratamiento de la enfermedad renal crónica», que ha sido patrocinado por AstraZeneca.
Conflicto de interesesJLG ha participado en advisory boards de Boehringer-Ingelheim, AstraZeneca, Bayer y Novo Nordisk; ha impartido conferencias para AstraZeneca, Boehringer-Ingelheim, Esteve, Bayer, Eli Lilly and Company, Astellas y Novo Nordisk, y ha recibido ayudas para la investigación de AstraZeneca.
VPC ha recibido subvenciones/apoyo a la investigación: FIS ISCIII-AES-2018/003116 (ESCARVAL-Genero); Grant AZ Agora Study, ESR-17-12871 (fundación SEMERGEN), así como honorarios por consultorías/ponencias de Laboratorios Servier, MSD, Daiichi-Sankyo, Novartis, Almirall, Sanofi, Pfizer, Viatris, Boehringer-Ingelheim, GSK, AstraZeneca, Esteve, Menarini, TEVA, Ferrer, Rovi, Vifor Pharma, VISO.
CG no declara conflicto de intereses.
